Historical Perspective of High-Throughput Screening
High-Throughput Screening (HTS) is one of the main sources of leads for drug discovery; the process involves evaluating a large diverse sample collection in an effort to identify compounds that possess a desired activity. In practice this resembles looking for a needle in a haystack since the vast majority of compounds in the sample collection will be devoid of the desired activity. Despite the seemingly unfavourable odds random screening remains an extremely valuable source of starting points DOI.
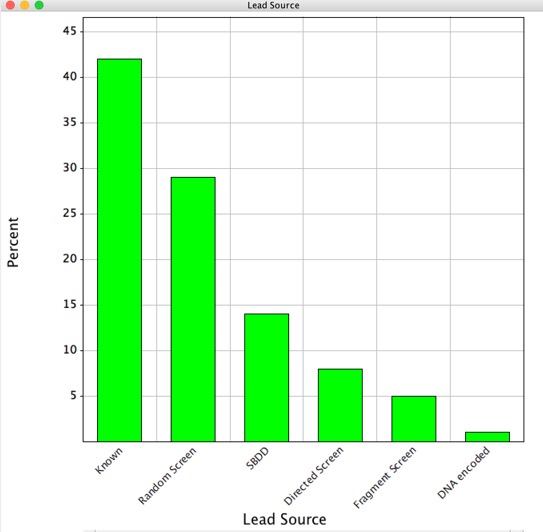
The products of this activity are referred to as “Hits”, once the activity has been confirmed to be appropriate with respect to potency/selectivity, the chemical structure and purity established, and preliminary ADME completed, these hits are often then described as “Leads”. In reality advances in robotics have resulted in the actual screening being relatively rapid whilst the subsequent follow-up is often the slow (and expensive) step, thus it is essential all “Hits” are genuine and have a high probability to become acceptable “Leads”. It is important to note that the function of the HTS is not to identify every possible hit from all compounds that are potentially available, rather it is to identify sufficient leads to support a drug discovery program and to prosecute this in the most timely and cost effective manner possible. With this in mind the quality and diversity of the screening collection is of critical importance.
Building up a sample collection for High-throughput screening is a major undertaking and for a small company or academic group submitting a proposal to the European Lead Factory might be an attractive alternative.
Factors effecting Screening Collection design
Physicochemical Properties
The lack of productivity in the Pharmaceutical Industry lead to a re-evaluation of HTS initially resulting efforts by companies to dramatically increase the size of the screening collection by employing combinatorial chemistry to prepare vast libraries of compounds. However this effort was largely unsuccessful and merely served to increase the size of the haystack rather than the number of needles. The seminal work by Chris Lipinski (Advanced Drug Delivery Reviews 46 (2001) 3 – 26) highlighted the importance of physicochemical properties in drug design leading to the “Rule of Five”. This states “In the discovery setting ‘the rule of 5’ predicts that poor absorption or permeation is more likely when there are more than 5 H-bond donors, 10 H-bond acceptors, the molecular weight (MWT) is greater than 500 and the calculated Log P (CLogP) is greater than 5.” Since then a number of other studies have made similar observations that attrition in the drug discovery process is linked to physicochemical properties of the candidate compounds.
Most recently a study of preclinical toxicity (Bioorganic & Medicinal Chemistry Letters 18 (2008) 4872–4875), found, across a wide range of chemical space, a greatly increased likelihood of toxicity was found for less polar, more lipophilic compounds. This results are perhaps not surprising when one considers the structure-activity relationships (SAR) of a variety of potential liabilities:-
- HERG, large lipophilic active site
- Partition into liver enhanced by increased lipophilicity
- CYP3A and CYP2D6 inhibition increased by lipophilicity
- CP450 Induction, PXR binding site is large and hydrophobic
- Plasma Protein Binding, affinity increases with logD
With this in mind there have been several studies evaluating the changes in molecular properties in going from the initial lead to the drug candidate (J. Chem. Inf. Comput. Sci. 2001, 41, 1308-1315) leading to the following observations.
“The following differences were observed between the medians of drugs and leads: ∆MW = 69; ∆CMR = 1.8; ∆RNG = ∆HAC =1; ∆RTB = 2; ∆CLogP = 0.43; ∆LogD74 = 0.97; ∆HDO = 0; ∆DFPS = 0.15; ∆PPFS = 0.12. Lead structures exhibit, on the average, less molecular complexity (less MW, less number of rings and rotatable bonds), are less hydrophobic (lower CLogP and LogD74), and less druglike (lower druglike scores). These findings indicate that the process of optimizing a lead into a drug results in more complex structures”.
Thus since lead optimization invariably leads to an increase in molecular complexity, molecular weight, and lipophilicity it is highly desirable to weight the screening collection towards smaller, less lipophilic compounds, however this should not mean that an interesting molecular structure of Mol Wt 501 should be excluded.
Toxicophores
Idiosyncratic drug reactions (IDRs) are a major concern for drug development, they often are not apparent in preclinical toxicity testing and are observed only when the drug is given to large patient populations. The postulated mechanism involves metabolic activation leading a reactive metabolite which can lead to thiol depletion (reaction with glutathione), reversible protein modification (glutathionylation and nitration), further irreversible protein adduct formation and subsequent irreversible protein damage leading to an immune mediated adverse event (Drug Discovery Today Vol. 8, No. 22 November 2003). Similar metabolic activation has been proposed to be responsible for the genotoxicty of some compounds and a variety of toxicophores proposed (Journal of the Iranian Chemical Society, Vol. 2, No. 4, December 2005, pp. 244-267).
Interference with the detection technology
In addition the new screening technologies have introduced new requirements, many now use fluorescence-based technologies and there is a need to avoid compounds that might interfere with the detection technology, or compounds that might chelate Calcium ions. Also some molecules can aggregate under the assay conditions giving misleading results, more information here.
A number of major pharmaceutical companies have devoted significant resources to evaluating their sample collection in order to identify a “screening collection”. Whilst the precise filters used by each company are confidential Oprea et al (J. Comp. Aid. Mol. Des. 14 (2000) 251-264) have described a “Leadlike” filter.
Frequent Hitters, False Positives, Promiscuous Compounds
Within any screening campaign there are a number of compounds that on further investigation fail to reproduce the initial activity in subsequent assays. There have been a number of studies trying to elucidate the structural features that would allow identification of these artifactual hits before significant effort has been expended following them up, this work is more fully discussed here.
Suggested criterion for inclusion in the screening collection.
For inclusion in the Screening Collection a molecule can have at most one violation of the following conditions:
1. The number N or O that are hydrogen bond donors must be 5 or less
2. The number of N and O atoms must be 8 or less
3. The molecular weight must be 150 to 450
4. The logP must be in the range [-3.5 to 4.5], inclusive
5. The number of rings of size three through eight must be 4 or less
6. The number of rotatable bonds must be 10 or less
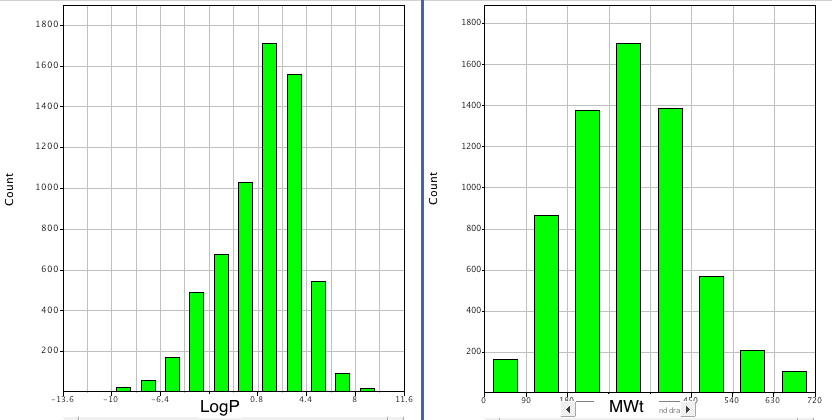
Whilst these provide useful guidelines it is worth looking at the profile of compounds taken from Drugbank, and it certainly looks like in terms of size and lipophilicity the guidelines are correct.
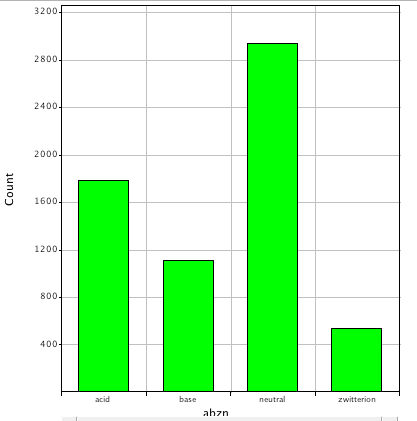
However one feature that seems to be constantly overlooked is the presence of ionisable groups. Looking at Drugbank again it is clear that more 50% of the molecules are predicted to be ionised at physiological pH.
Ionisable groups have an important role in reducing LogD, improving solubility and pharmaceutical properties by allowing a range of options in salt selection. Yet it seems "diversity" in screening collections is often achieved by simply capping a range of amines with a wide range of acid chlorides or sulphonyl chlorides, resulting in collections that are seriously compromised with respect to ionisable groups.
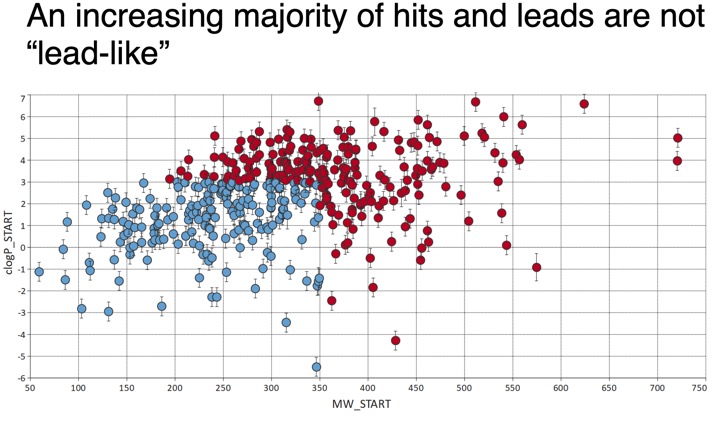
However it should noted that emerging areas include protein-protein interactions and epigenetic targets both target areas include many with open shallow binding sites requiring lager molecules to achieve high affinity binding, for these targets it may be necessary to widen the screening range. Recent work look at hits for new targets suggests the starting hits are getting larger.
In addition the molecule should not contain any of the following reactive groups/toxicophores:-
The molecule must not contain groups in a conservative list of reactive groups: acrylamide, acyclic diketyl, acyl halide/cyanides, activated esters, cyclohexadienes, aldehyde, aliphatic imine, aliphatic ketone, aryl or aliphatic nitro(so)/hydroxylamine, aliphatic (thio)ester, alkyl halide, anhydride, azide, aziridine, beta-heterosubstituted carbonyl, epoxide, halopyrimidine, hetero-allyl, iso(thio)cyanate, maleimide, Michael acceptor, perhalo ketone, phosphonate ester, phospho-, sulfonate ester, thiol, disulphides, thio(urea), O-O single bonds and transition metals.
There are a number of commercial software tools for filtering compounds, there is also the open-source Filter-it built on OpenBabel by Silicos-it. Filter-it™ is a command-line program for filtering molecules with unwanted properties out of a set of molecules. The program comes with a number of pre-programmed molecular properties that can be used for filtering (Leadlike, Druglike, etc. ) users can of course develop their own filters, full details are available online. There are also a number of published filter scripts for tools like Vortex.
Diversity
A structure search would need to be performed to ensure each sample is unique and, depending on the number of samples, a diversity analysis may need to be performed to avoid the collection being biased towards a particular chemotype. Typically any clusters of 10 or more compounds with >85% similarity should be examined to reduce the size of the cluster, an exception may be focussed screening libraries. Deliberately building clusters around privileged structures is certainly worth thinking about.
See Also Focused Screening Libraries
Natural products have historically provided key hits for a significant number of programmes, and whilst they may not meet the inclusion criteria described above, there is certainly a case for maintaining a collection for screening for anti-infective targets.
Ongoing Activities
It should be recognised that the screening collection will not be a static entity and will need to be continuously updated and annotated.
- Samples will be depleted with use and a decision will need to be made as to whether they should be replaced.
- Novel chemotypes may become available, these would need to pass the lead-like filters above and be dissimilar to existing compounds.
- As screens are run promiscuous compounds will be identified and flagged and possibly be removed.
- New screening technologies may be compromised by some chemotypes.
- Compounds followed up as leads may reveal unexpected toxicity, this information should be captured and the sample information annotated.
Bayer have described in detail their efforts to enhance their screening collection "An approach towards enhancement of a screening library: The Next Generation Library Initiative (NGLI) at Bayer — against all odds?" DOI they note that " over time, increasing, efforts were generally required to reach novelty when starting with hits from our HTS collection". This is due to a combination of factors, internal projects by their nature focus on a limited number of hit chemotypes, in addition acquisition campaigns are largely nonexclusive, commercially available compound sets. They found that 18% of all compounds in their library were commercially available, and 56% were similar to compounds in PubChem. This "novelty erosion" was the key driver for their library expansion initiative.
They set the following criterion:
- Molecular properties (average): oral phys-chem score (oPCS, DOI) <2, MW <400, calc logD7.5 <3.0, Fsp3 >0.4.
- Novelty: scaffold and/or substitution pattern should provide two points of deviation from literature precedence.
- Synthetic tractability: maximum of 10–12 linear steps, plausible synthetic scheme. Structural features: no unwanted functionalities
They used different approaches depending on the target class, for GPCR and Ion channels they used random forest models based on ChEMBL data to help select new molecules. For proteases they used the vast amount of co-crystal structures to set up an automated docking and structure-based design approach. The Kinase design team primarily designed new cores and prioritised those by docking or used ligand-based pharmacophores. They complemented this design process by including libraries of peptidic or non-peptidic macrocyclic ligands.
Quality Control
Ideally the samples should have a single major component (>95%), and sufficient sample should be immediately available for follow up.
The quality and purity of the screening collection can have an enormous impact on resources when one considers the time wasted following up impure, degraded or incorrectly assigned hits from screening. There are no hard numbers but I’ve heard of cases where up to 40% of hits failed to provide a useful lead due to the poor quality of the screening sample.
Of course QC of a million compound screening library is a significant drain on resources, some companies adopt a strategy of evaluating a random selection on a regular basis, others the entire collection. In both cases, because the vast majority of compounds will never be hits in an assay, resources are being wasted doing QC analysis on “inactive” compounds.
Perhaps a better strategy is to adopt an “In and Hit” protocol, compounds are rigorously evaluated before they are added to the screening collection and then only reassessed if they are a hit in a screen. Any compounds that fail QC as a hit are then removed from the collection. The degree of degradation is very much dependent on the storage conditions and there have been several publications evaluating different conditions.
Ideally the initial quality control should asses both purity and structural characterization. Samples would be expected to be a single major peak (>95%) by HPLC using two different columns, they would should also have the correct mass. Most of which can be automated. Compounds should also be analysed by 1H NMR however this probably requires a greater degree of human interaction and it may be only possible to do this for the “Hits”.
Metal impurities are becoming more common with the use of metal mediated cross-couplings etc. whilst these would not be detected by typical HPLC, NMR or Mass Spec they would be observed in a CHN analysis however automated synthesis techniques usually means too little is prepared. Metals can be removed by the correct workup, e.g. ammonia solution for Cu, Charcoal, and there are a variety of scavenging silicas.
If you are building up a collection from commercial suppliers then it is worth reading the Chemical Databases and Suppliers page. However it is essential to check the identity and quality of samples from external vendors, a communication from the Broad Institute highlights the potential problem.
Surprisingly, 2,482 of 8,584 samples (29%) failed QC, defined as a purity of less than 85%, as measured by UPLC absorbance peak area at 210 nm, or by an evaporative light-scattering detector (ELSD) for peaks containing the expected compound mass
Worth reading:-
Stability Through the Ages: The GSK Experience, Journal of Biomolecular Screening, Vol. 14, No. 5, 547-556 (2009)
COMDECOM: predicting the lifetime of screening compounds in DMSO solution , Journal of Biomolecular Screening, Vol. 14, No. 5, 557-65 (2009).
Broad Coverage of Commercially Available Lead-like Screening Space with Fewer than 350,000 Compounds, Jonathan Baell DOI
Metal Impurities Cause False Positives in High-Throughput Screening Campaigns, DOI
An approach towards enhancement of a screening library: The Next Generation Library Initiative (NGLI) at Bayer — against all odds?" DOI
Is there a difference between leads and drugs? A historical perspective. J Chem Inf Comput Sci. 2001 Sep-Oct;41(5):1308-15 DOI.
Updated 25 November 2019