Deconstruction of a screening hit
The chemical starting point for a drug discovery programme can come from a variety of different sources this could be a single known compound or multiple different series from a HTS campaign. One strategy is to look for similar compounds from commercial sources for hit expansion, sometimes called SAR by catalogue. Another approach is to take the initial lead and look to simplify it with the aim to identify the minimum essential pharmacophore.
In our work on NK1 antagonist the quinuclidine structure below appeared in a Pfizer patent, whilst very potent this had a number of liabilities, it had high LogP and negligible bioavailability. The quinuclidine scaffold also presented significant synthetic challenges.
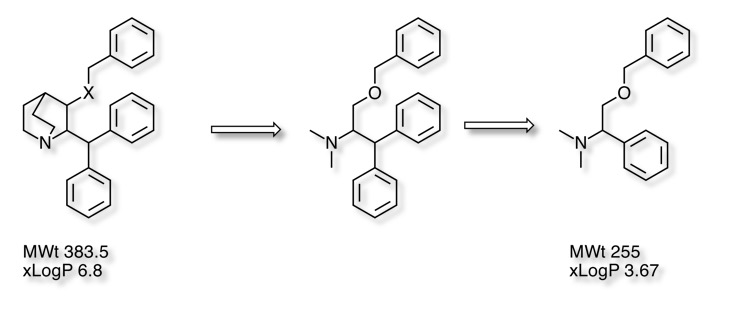 Our strategy was to simplify the structure by removing the bicyclic structure to give and acyclic system and subsequent removal of one of the phenyl rings This gave us the minimum pharmacophore . Whilst lower in affinity the result was lower molecular weight and the LogP reduced by 3 units. Indeed the molecule now occupied "fragment space". When I presented this at the recent Fragments meeting a member of the audience coined the phrase "Deconstruction of a screening hit" and several people used the phrase subsequently so perhaps it will used in the future.
Our strategy was to simplify the structure by removing the bicyclic structure to give and acyclic system and subsequent removal of one of the phenyl rings This gave us the minimum pharmacophore . Whilst lower in affinity the result was lower molecular weight and the LogP reduced by 3 units. Indeed the molecule now occupied "fragment space". When I presented this at the recent Fragments meeting a member of the audience coined the phrase "Deconstruction of a screening hit" and several people used the phrase subsequently so perhaps it will used in the future.
With the minimum pharmacophore in hand, we and others embarked on a series of approaches to introduce conformational constraints, shown below. All of these approaches led to sub nanomolar affinity molecules.
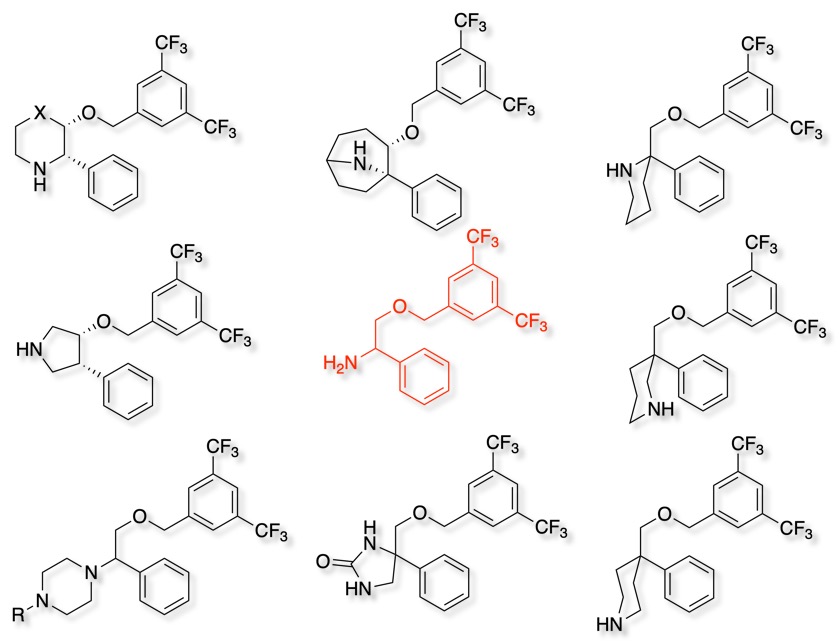
I do wonder how many of these different scaffolds would have been explored if the initial deconstruction of the original lead had not been conducted?
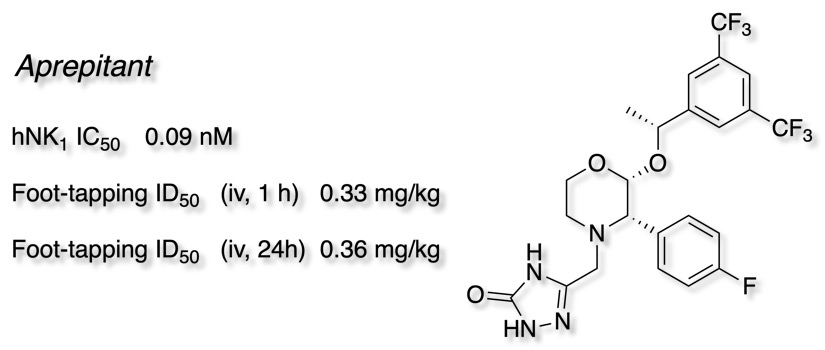
This work culminated in the identification of Aprepitant (Emend).
Updated 8 April 2024