Fragment-Based Screening
Fragment-based screening has become increasingly popular over the last 15 years and has proven to be a viable alternative to high-throughput screening. The appeal has been driven by several features
- “Fragment Space” is smaller than “Chemical Space” and can be more effectively probed with a relatively small library
- A million compounds cover only a small fraction of the suggested 1060 Chemical Space, whilst 2000 compounds can probe much of the 106 Fragment Space
- Protein requirements should be smaller
- Binding Efficiency for small molecules is likely to be higher
- Hit rates for Fragment-based screening appear to be higher, typically 3-10%.
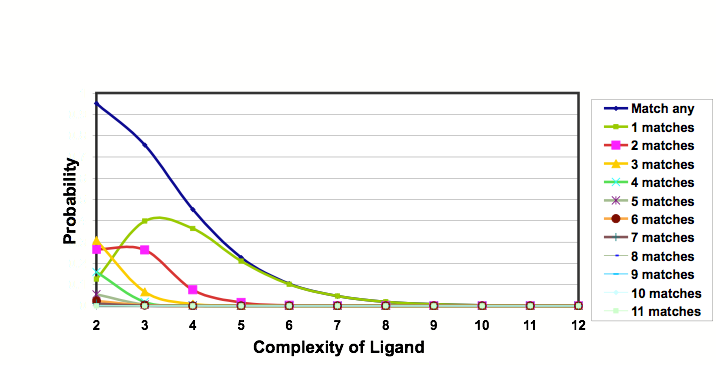
Theoretical studies by Hann using a simple model of ligand-receptor interactions, in which the interactions between ligands and receptors of varying complexities are studied and the probabilities of binding calculated. It was observed that as the systems become more complex the chance of observing a useful interaction for a randomly chosen ligand falls dramatically. In addition the process of optimizing a lead into a drug results in more complex structures, suggesting the attraction of less complex starting points. Similar conclusions were uncovered by Opera.
Probabilities of ligands of varying complexity matching a binding site of complexity 12
+-+-+-+-+-+-+-+-+-+-+-+- Binging Site +-+-+-+-+-+-+-+-+-+-+-+-
-+-+--+++----++--+-+ Ligand +-+-+
As the ligand/receptor match becomes more complex the probability of any given molecule matching falls to zero.
But less complex ligands will have lower affinity
Screen less complex molecules to find more hits
Less potent but higher chance of getting on to the success landscape Opportunity for medicinal chemists to then optimise by adding back complexity and properties, computational tools are now being developed to support expanding fragment hits. Requires much more sensitive assays than generally used in high-throughput screening
Need for it to be the right sort of molecules.
Screen at up to 1mM so solubility can be an issue, some fragment collections now include measured solubility this should be regarded as a significant advantage. Aggregation at high concentrations can lead to false positives. It may be useful to use two different screening technologies and then focus on compounds that are active in both assays.
Screening Methodologies
Bioassays
If the assay is robust in the presence of high concentrations of ligand then bioassays provide a rapid and quantitative methods for detection requiring little protein. In practice however many assays prove unsuitable, with false positives as a result of compound aggregation at the concentration of ligands used, interference with assay end point (e.g. optical interference fluorescence, quenching, toxicity, etc.) or disruption of the protein by unfolding, disruption of protein complexes or precipitation. Additionally, false negatives can occur due to lack of effective solubility of compounds in assay buffers. Whilst in theory any bioassay could be used for Fragment-based screening they often lack the sensitivity to identify weakly binding fragments. Developing high-throughput screens Ion channels has traditionally been challenging and so it was interesting to note a recent publication describing the development of a medium-throughput automated patch clamp (PatchXpress, (PX); Molecular Devices, Inc., Sunnyvale, CA) assay (DOI : 10.1016/j.bmcl.2010.12.115 ) used for screening fragments against acid-sensing ion channel 3 (ASIC3).
BLI
A recent paper has described the use of Biolayer interferometry as a fragment screening technique. BLI is a methodology that measures changes in an interference pattern generated from visible light reflected from an optical layer and a biolayer containing proteins of interest. Like SPR Biolayer interferometry requires the protein to be imobilised on a solid support, the authors describe several screens and completed a screen of 6500 fragments in 10 days using 1mg of eIF4E protein. Primary hit rates with BLI are target dependent and were 21, 24, and 3.5% for PPI targets BCL-2, JNK-1, and eIF4E, respectively. Approximately 50% of the hits confirmed in biochemical assays. It has been suggested that BLI like SPR may be prone to high rates of false positives.
CE
The use of Capillary Electrophoresis in fragment-based drug discovery has been pioneered by Selcia. CE is a high-resolution technique that can detect both high and low-affinity molecular interactions. The advantages are very low protein consumption, no need for highly pure protein, no protein size limit and you doesn’t need to worry about the effects of immobilization or conjugation, it is solution based, involving a separation step: insensitive to aggregation phenomena and has a low false hit rate, this technique generates IC50 data. However it does not offer the insight into the docked ligand that X-Ray might. This technology has also been used to look at protein-protein interactions, and a paper has been published describing identification of HSP 90 ligands DOI.
19F NMR
Whilst NMR based screening is well established the use of 19F NMR has recently generated significant interest. In particular a recent publication Zhong et al describes the construction of a 19F fragment library based on the traditional fragment “rule of three” with comparable size and chemical diversity to a typical fragment library (∼1k−2k compounds). 19F NMR experiment (with 1H decoupling) is significantly faster and, in many ways, more robust than traditional 1H-based NMR screening
ITC
Isothermal titration calorimetry (ITC) is rarely used for fragment based screening but a recent paper by Ladbury et al has highlighted the potential. In theory two samples are mixed together, and the change in heat is precisely measured. If one solution contains a protein and the other a small molecule, one can determine the enthalpy (deltaH) as well as the overall free energy (deltaG) of binding (and thus the affinity). In practice fragment interactions may be too weak for detection and masked by technological challenges. It does however provide an insight into the enthalpy changes which may be a more attractive route for optimisation rather than rely on entropic effects.
MS
The use of mass spectrometry has been reviewed DOI. Under careful conditions protein-small molecule complexes can be observed. The process requires very small amounts of protein and you obviously doesn’t need to worry about the effects of immobilization or conjugation. However the required conditions (lack of detergent) can lead to false positives due to aggregation.
MST
Microscale Thermophoresis (MST), is described in a paper in Angew Chem DOI. It makes use of the intrinsic fluorescence of proteins to quantify the binding affinities of ligands and discriminate between binding sites. It uses native protein,and you doesn’t need to worry about the effects of immobilization or conjugation. However it relies on the fluorescence signal of native amino acids and so could be obscured by some ligands.
NMR
NMR has been used on a number of occasions and has the advantage of generating binding affinities for even weak ligands. However NMR also requires a relatively large amount of protein and not all proteins are of the required size/solubility. Target-based NMR screening monitors the changes in 1H,13C or 1H15 N correlation signals for a labeled protein,when binding to a test compound. The method can detect nM to mM interactions and provides structural information of the binding site. However, it normally requires large quantities of isotopically labeled protein (50 – 200 mg), with protein solubilities between 0.1 and 1 mM. Ligand-based NMR screening relies on changes in the ligand signals when binding to the target, it does not require labeled protein, and the protein consumption is substantially lower. It is possible to screen carefully chosen mixtures of compounds Computer-Aided Design of Fragment Mixtures for NMR-Based Screening.
Video provided by Bruker
SPR
Surface Plasmon Resonance is known to be a powerful tool for studying biomolecular interactions in a sensitive and label-free detection format, it is possible to evaluate larger libraries of compounds but either protein or ligand must be immobilized. Graffinity use a small molecule library consists of 20,000 fragments and 90,000 lead-like compounds immobilized on chemical microarrays. Kinetic Discovery currently offers services using either their own specially selected fragment library or a customer's collection and there is a recent publication describing the methodology.
Thermal Shift Assay
Monitoring changes in protein stability has become a regularly used method to identify fragments, as the temperature rises the protein unfolds and exposes the hydrophobic core, this can be measured by binding to a fluorescent marker. Ligands that bind to the protein would be expected to increase protein stability. DOI
Weak Affinity Chromatography
Weak affinity chromatography has recently been used for fragment screening DOI. This technique represents a simple and robust procedure and it is based on weak zonal affinity separations of analytes such as small molecules. In the example described in the publication thrombin was immobilised on a silica column and in order to increase throughput mixtures of compounds were added with the mass spec detection.
X-Ray
The use of X-ray crystallography as the detection technique has now been demonstrated on numerous occasions following the pioneering work by Astex. A clear advantage of this approach is that false positives are reduced because if a compound is seen by crystallography it is possible to evaluate the validity. In addition it may be possible to assess how to enhance binding by using modeling techniques or purchase of related analogues. On some occasions different ligands may occupy different volumes of the binding site which may allowing “linking” to enhance binding affinity On the other hand, these techniques can be very time and resource intensive in which a large (Crystallography requires 10-50 mg of target protein with a purity of > 95%) protein construct is needed, is compatible with crystallisation to yield robust crystals and diffracts well and is compatible with ligand binding (on occasions the ligand binding pocket may be on the interface between two monomers in the unit cell obstructing ligand access). Also fragment ligands being considered will need to be soluble in the crystallisation medium. No affinity information is obtained from a crystallographic experiment.
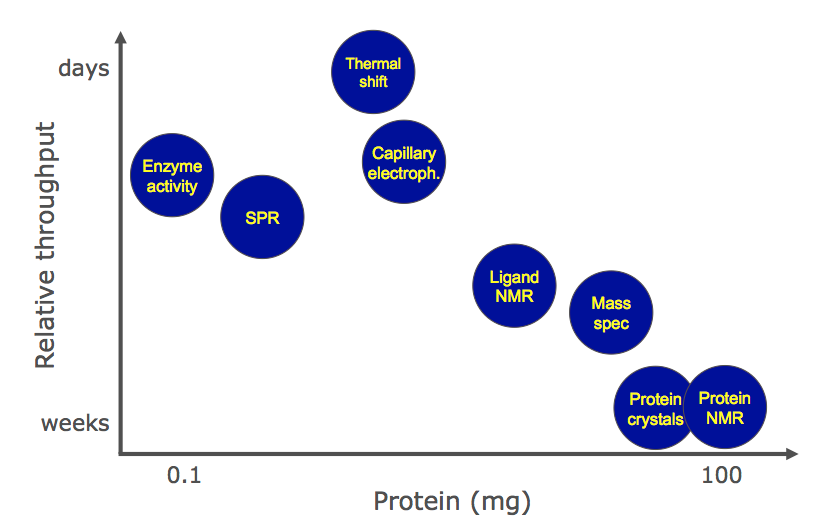
When comparing the different technologies this slide shown by Al Gibbs at the Fragments 2013 meeting is perhaps useful.
Companies offering fragment screening services
Astex X-ray, NMR, ITC 1,600 compound library
Alveus Pharma "Details coming soon"
Beactica SPR 1946 compound library
BioFocus SPR 1,500 compound library, also offer X-ray and Mass Spec
Biosensor Tools SPR 1500 compound library
Carmot Therapeutics Chemotype evolution (tethering)
Crelux Microscale thermophoresis X-ray 1,000 compound library
Crown Biosciences X-ray, SPR X-ray 3,400 compound library
CrystalsFirst X-ray, 1000 compound library, or clients library, in house technology improves efficiency and number of hits
Emerald BioStructures X-ray, NMR 1,500 compound library
Evotec Fluorescence correlation spectroscopy SPR, NMR, X-ray, biochemical 30,000 compound library
IOTA Pharmaceuticals SPR, biochemical, and NMR X-ray 5,500 compound library
Kinetic Discovery SPR 700 compound library
Kinomed X-ray
Novalix SPR array MS, NMR 24,000 compound library
Pharma Diagnostics Bead-based SPR compound library
Polyphor SPR 1,000 compound library
Proteros TR-FRET X-ray 8,000 compound library
Selcia Capillary electrophoresis 1,300 compound library
Sprint Bioscience X-ray,
Structure Based Design X-ray, 1000 compound library
Vernalis NMR, biochemical, SPR, ITC X-ray 1,400 compound library
Viva Biotech X-Ray, NMR, SPR 2000 compound library
XChem at Diamond X-ray, 700 compounds per day
Zenobia X-ray, SPR X-ray 1,000 compound library
ZoBio TINSa NMR, SPR 1,500 compound library
A possible strategy for fragment-based screening
A number of groups have published finding suggesting X-ray success rate is low (~25-30%) when hits unique to a single screening method is pursued. However confirmation of hits by at least one other screening technology greatly improves the success rate, given the resource implications for committing hits for X-ray determination a better strategy might be.
- High capacity screen
- Confirmation of hits in orthogonal screen
- SAR by catalogue (5-10 compounds)
- Biochemical confirmation
- Structural studies on interesting series (X-Ray)
- Structure-based design
Having discovered one or more fragment hits, the hit validation stage of a project involves testing related analogues of a fragment hit. The purpose of this phase is to understand the nature of binding more fully, to generate some interpretable structure activity relationships (SAR) around the hit, and to optimize the fragment itself before it is grown toward a lead compound. If close analogues of a fragment are available in-house or commercially, this often allows for quicker and cheaper hypothesis testing than synthesis of new compounds. Hence “SAR by catalogue/collection” is a popular way to rapidly explore the chemical space around a fragment. This will obviously have implications for "bespoke" fragments for which there may be no readily available fragments. Identifying "similar" fragments can be a challenge because many of the fingerprints used in similarity searches are based on conventional drug-like molecules (MW 350-500) and so the fragment fingerprints can be very sparse. A matched molecular pair analysis allows compounds that differ by a single point of change to be retrieved. Astex have described The Fragment Network: A Chemistry Recommendation Engine Built Using a Graph Database DOI, a graph based database and user friendly interface.
Another approach is to have easily exploitable synthetic chemistry handles on the fragments (e.g. aryl halides) that allow rapid exploration, or established late-stage modification chemistry in place.
Ligand Efficiency
One of the principle attractions of fragment-based drug discovery has been the observation that the smaller ligands bind more efficiently when comparing of the binding affinity per heavy atom
LE= -RTln(IC50)/HAC
LE = (ligand efficiency)
HAC = (Heavy Atom Count)
More recently Lipophilic Ligand Efficiency
LLE = pKd-logP
or LLE = pKd-logD
has been used or even a combination, however this metric is not molecular weight adjusted and so LELP has been proposed to be a better metric
LELP = logP / LE (where LE = ligand efficiency)
Which does account for molecular weight. A comparison of the different metrics has been carried out DOI: 10.1021/jm201388p and conclude LELP is probably the better metric to use for fragments.
LLE is sensible for the development stages but does not prefer fragment-type hits, while LELP has an advantage for this class of compounds and discriminates preferred starting points effectively. Both LLE and LELP have significant impact on ADME and safety profiles; however, LELP outperforms LLE in risk assessment at least on the present data set.
Building from Fragments
Whilst fragments provide very attractive hits from screening they usually have only modest affinity, several approaches have been described to optimize the structures. Recently Sarti et al have described experimental work looking at the entropic influence of a linking strategy in fragment-based drug design. In particular they looked at the effect of linking two weakly binding fragments, in which an increase in affinity of 3 orders of magnitude are observed. The difference in TΔS for the binding of the linked ligand compared to the fragments is high and negative (-4.30 kcal/mol) and fully accounts for the increase in affinity.

“This additional increase in affinity can be explained by considering that when two ligands bind to a protein, each of them loses a fraction of rigid body rotational and translational entropy. Conversely, when the two ligands are tethered in a single molecule, only one unfavorable rigid body entropy barrier affects the binding.”
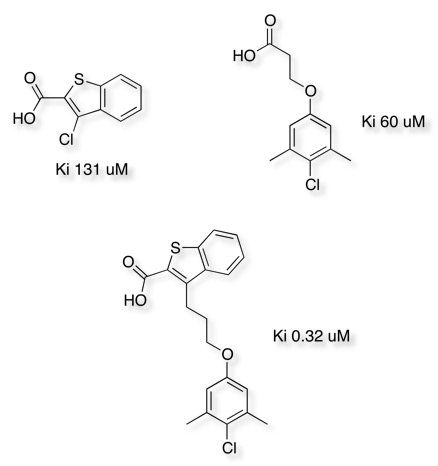
The impact of linking two chemically distinct fragments can also be seen in the work towards Potent Myeloid Cell Leukemia 1 (Mcl-1) Inhibitors DOI shown below.
Abell et al have compared the cross-linking, with fragment growing approach, looking at Inhibitors of Mycobacterium tuberculosis Pantothenate Synthetase.
“The two strategies resulted in similar compounds with similar potencies. This outcome obscures the fact that although the linking strategy appears more elegant, the limited repertoire of linkers is likely to compromise the binding of the original fragments. In comparison, the fragment-growing strategy provides more freedom for development at each stage and allows more room for further optimization.”
A general consensus from scientists working in the area is that the key to successful prosecution of fragment-based drug design is the availability of structural information, so whilst there are a steadily increasing number of technologies available for the initial screening of fragments generating the protein crystal structure is still a critical and often rate-limiting step in the process. Diamond provide a tips and tricks page based on their (and SGC) experience Tips and tricks. It might also be worth reading "High-throughput production of human proteins for crystallization: The SGC experience" DOI and "Lessons from high-throughput protein crystallization screening: 10 years of practical experience" DOI. The Practical Fragments Blog is an invaluable resource for news in the area of fragment-based drug discovery, highlighting relevant conferences and publications with commentary and discussion from experts. They also maintain a list of compounds now in the clinic that were discovered using fragment based screening. To date the list has > 30 entries with the first, a B-Raf enzyme inhibitor Vemurafenib and Venetoclax for the treatment of CLL now approved.
There are now a few books available describing Fragment-Based Drug Discovery (links to Amazon)
Fragment-Based Drug Discovery: Lessons and Outlook (Methods and Principles in Medicinal Chemistry)
Fragment-based Drug Discovery: A Practical Approach
Fragment-Based Approaches in Drug Discovery (Methods and Principles in Medicinal Chemistry)
Worth Reading:- Special Issue: Fragment-Based Drug Design / Guest Edited by D. Joseph-McCarthy, Journal of Computer-Aided Molecular Design Volume 23, Number 8 / August, 2009 There is an entire issue of Current Topics in Medicinal Chemistry Dedicated to Fragment-Based Drug Discovery Volume 9, Number 18, December 2009.
Updated 4 March 2021