Human Ether-a-go-go-Related Gene (hERG) Blockers
What is the issue?
- In recent years, a number of drugs have been withdrawn from the market because of reports of sudden cardiac death.
- In all cases, long QT syndrome an abnormality of cardiac muscle repolarization that is characterized by the prolongation of the QT interval in the electrocardiogram, was implicated as inducing ventricular tachycardia that can spontaneously degenerate to ventricular fibrillation and cause sudden death.
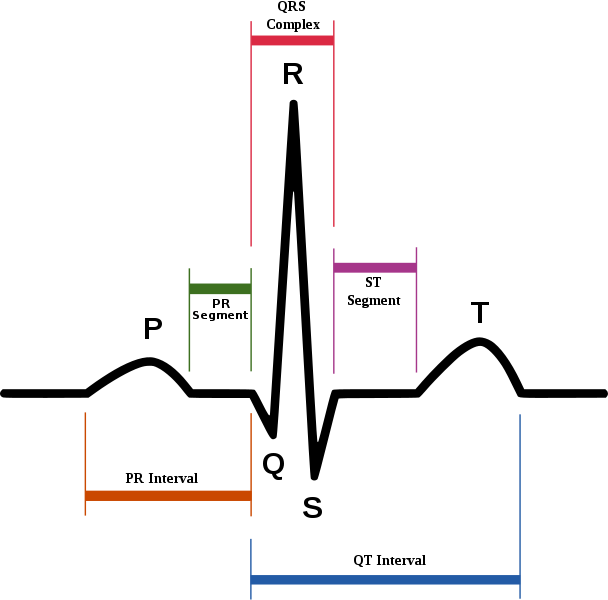
- Virtually every case of a prolonged duration of cardiac action potential related to drug exposure can be traced to one specific mechanism: blockade of IKr current in the heart.
- Blockade of Kv11.1 potassium ion channel encoded by the hERG gene (KCNH2) is widely regarded as the predominant cause of drug-induced QT prolongation. This channel is critical for the repolarisation of the cardiac membrane. hERG forms the major portion of one of the ion channel proteins (the 'rapid' delayed rectifier current (IKr)) that conducts potassium (K+) ions out of the muscle cells of the heart, and this current is critical in correctly timing the return to the resting state of the cell membrane during the cardiac action potential.
- If drug levels of even a modest affinity hERG ligand rise, due to inhibition of a co-administered CYP inhibitor, QT prolongation may be observed
- Since 2005, the FDA has required that new molecular entities are evaluated in a Thorough QT (TQT) study to determine a drug's effect on the QT interval. The TQT study serves to assess the potential arrhythmia liability of a drug. As the pharmaceutical industry has gained experience in performing TQT studies, it has become evident that traditional QT correction formulas such as QTcF, QTcB, and QTcI may not always be suitable for evaluation of drugs impacting autonomic tone.
In 2005, the International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use (ICH) S7B guidance lays out a non-clinical testing strategy to assess cardiac risk with two components. The first is an in vitro assay of the current through the HERG (also known as KCNH2) potassium channel — which is a major contributor to the repolarisation phase of the cardiac action potential — as its inhibition is the most common mechanism by which drugs prolong the QT interval. The second is an in vivo QT assay in an animal model. The ICH E14 guidance details the testing of a drug’s effects on healthy volunteers’ QT interval (a thorough QT study), with a mean measurement of more than 5 milliseconds above normal serving as a threshold for regulatory concern.
Typically, a HERG functional current assay costs about US$20,000, and an in vivo assay in dogs can cost $100,000, In addition the cost of a thorough QT study in humans depends on the size of the study but can reach several million dollars. In addition rabbit and dog ventricular wedge studies may also be required.
Typical Inhibitors

Pharmacophore model consists of a basic nitrogen center flanked by aromatic or hydrophobic groups attached with flexible linkers, receptor modeling and mutagenesis studies highlight interactions between ligand (MK-499) and V625, G648, Y652 and F656. PNAS 97, 12329 (2000) are consistent with the pharmacophore model. The pharmacophore model from J. Med. Chem. 2002, 45, 3844-3853 DOI 10.1021/jm0208875 is shown below.

In a more recent study DOI using 18 chemically diverse hERG blockers from the literature, all evaluated with the same patch clamp assay identified similar features, a basic nitrogen (red), an aromatic ring (blue) and two further lipophilic regions (cyan). Whilst this was able to identify most of the actives in the test set it had a relatively high rate of false-positive hits, in addition when tested against an externally assembled data set the performance was lower. When the analysis was restricted to potent hERG blockers with IC50 values of ≤1 μM, the model was able to find 85% of the very potent ligands.
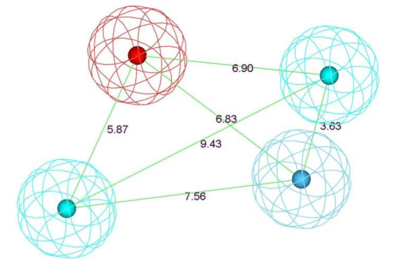
The authors also created a consensus model developed from 6 individual pharmacophore models (one of which did not contain a basic nitrogen), the consensus model was able to cover 84.9% of truly active compounds (ranging from 8 to 30% as single models) with a false positive rate of 3%. However once again the performance against an external data set was significantly poorer.
It should be noted however that non-basic HERG ligands are also known and Aronov and co have developed pharmacophore models for non-charged hERG blockers (J. Med. Chem., 2006, 49, 6917-6921.)
Empirical observations
- Zwitterions very rarely cause problems
- Almost all models include cLogP, a reduction of 1 logP unit in logP often leads to a 0.8 log unit in hERG activity
- Modify pi-Stacking interactions to reduce affinity
- Attenuation of pKa of basic amines, or steric blocking
- Do both, beta-alkoxy/hydroxy reduce pKa and LogP
- Acids less of an issue except for very lipophilic acids.
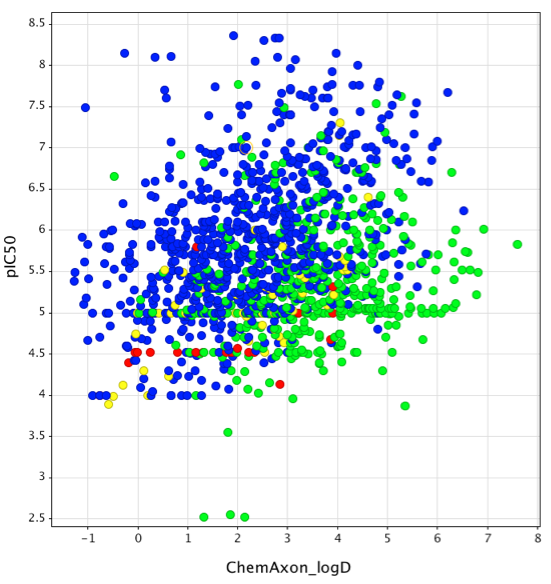
Given the issues that many drug discovery programs have had with off-target activity at the hERG channel I've started to compile a database of literature binding data. To date I have information on over 1600 compounds, my aim is to use the data to develop predictive models that I will make freely available, together with the complete data-set which I hope can be used to test novel models. The plot below (created using Vortex)shows pIC50 calculated from the literature IC50 data versus calculated logD determined using the ChemAxon tools, the colour coding shows the overall general tend of increasing hERG activity as logP increases with bases (blue) being more prone to HERG activity than neutral (green) for a given lipophilicity. It also highlights the reduced liability generally seen with acids (red) and zwitterions (yellow).
Strategies for Reducing HERG activity
Whilst there have been several attempts to build global models to predict HERG activity all seem to suffer a drop in performance when applied to chemical space outside that defined by the original training set. This has been explored in considerable detail DOI.
The work presented here shows the considerable variability in results that may be obtained by training and validating the same hERG blocker classifiers on different datasets. This is highlighted by the considerable reduction in model performance observed on our dataset with the models proposed by Dubus et al and by Thai and Ecker
Local models built using data for the chemical series under investigation would almost certainly out-perform any of the global models, however matched pair analysis by Willett et al DOI has highlighted a number of structural changes that may be of general interest. This has been extended by a fingerprint pair analysis by Springer et al
In the majority of cases substituents that increase lipophilicity (H to Br, H to CF3, H to Ph, Me to iPr, OMe to CF3) resulted in an increase in HERG binding. In contrast substituents that reduce lipophilicity (H to OH, Cl to CN, pyrrollidine to morpholine) reduce HERG binding
Replacement of H by OMe has different effects depending on the context, replacement of an aliphatic hydrogen by methoxy results in a greatly increased chance of reducing HERG activity, whereas the opposite effect is seen in the cases where the H to OMe transformation takes place next to an H-bond-accepting aromatic ring (such as pyridine or quinoline) resulting in increased chance of HERG binding. Replacement of H by OMe on a simple phenyl ring is largely neutral.
Introduction of an aliphatic oxygen near to an amine can significantly reduce HERG binding, as can be seen below this can be ascribed to a reduction on the pKa of the basic nitrogen, but also the hydroxyl serves to reduce the overall lipophilicity. It is worth noting that simply reducing the basicity of the nitrogen by introduction of F can actually have a detrimental effect since the lipophilicity of the molecule may actually increase.
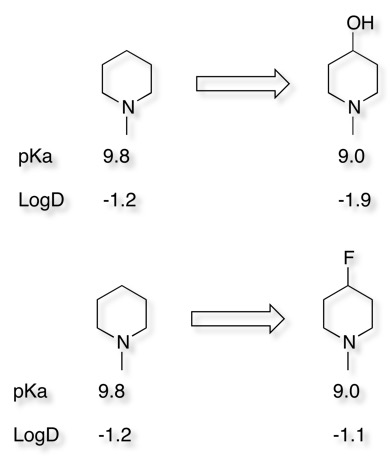
Changing a molecule with two amines into a system with one amine and a hydroxyl group reduces HERG inhibition
I undertook a matched pair analysis of the 1600 compound HERG dataset I’ve compiled from the literature using Vortex and a number of additional features were identified.
Introduction of a carboxylic acid almost invariably reduces HERG activity. In a couple of examples the acid was introduced at several different positions within the molecule and a reduction in HERG binding was observed irrespective of the position of the substituent.
Replacement of a chlorophenyl or fluorophenyl by pyridyl (a common bioisosteric replacement) results in a reduction in HERG binding, in fact introduction of (further) nitrogens into 5 or 6 membered heteroaromatic rings reduces HERG binding.
Acylation of basic nitrogens reduces HERG activity,
The effect of aromatic substitution is somewhat unpredictable but polar, electron-withdrawing substituents e.g. -CN seem to reduce HERG binding.
Functional Assay
Whilst the majority of data is derived from radioligand binding experiments (using either Dofetilide or MK-499), there is a substantial amount of data from patch clamp experiments, particularly using the ionworks system. I collated enough data now (covering 6 orders of magnitude) to give an idea of how the assays compare. As you can see there is a reasonable correlation between the assays, but there are one or two outliers. Which is more predictive of in vivo activity is an excellent question that I don’t have the data to answer yet.

I still need to increase the size of the data-set, and if anyone can direct me to any publicly available data, or to publications that contain data I'd much appreciate it.
What level of selectivity is needed?
In vitro binding affinity does not always correlate with observed in vivo activity (Some drugs that are not proarrhythmic, such as verapamil, phenobarbital and ranolazine, show positive results on a HERG assay. Verapamil, for instance, also acts on a calcium ion channel that cancels out the effects of its inhibition of HERG on the action potential Need to check early in programme and it is is better to focus in vivo on a window (10-100 fold) over the observed plasma Cmax at the likely clinical dose, but remember QT prolongation can be seen with low levels of HERG channel occupancy. An analysis by Redfern http://cardiovascres.oxfordjournals.org/content/58/1/32.full suggested that a ratio of more than 30 : 1 between hERG IC50 and the Cmax at therapeutic doses could be taken as indicative of a lack of TdP inducing potential.
The dataset confirms the widely-held belief that most drugs associated with TdP in humans are also associated with hERG K+ channel block at concentrations close to or superimposed upon the free plasma concentrations found in clinical use. A 30-fold margin between Cmax and hERG IC50 may suffice for drugs currently undergoing clinical evaluation, but for future drug discovery programmes, pharmaceutical companies should consider increasing this margin, particularly for drugs aimed at non-debilitating diseases.
It should be noted that it is likely that only the free drug can bind to the HERG channel so high plasma protein binding may influence the in vivo effects, and of course differences in protein binding between species may cause unexpected complications.
A study by Yao compared sixteen marketed drugs from various pharmacological classes with a known incidence—or lack thereof—of QT prolongation in humans, in hERG (human ether a-go-go-related gene) patch-clamp assay and an anaesthetized guinea-pig assay for QT prolongation DOI
Analysis of hERG inhibitory potency in conjunction with drug exposures and QT interval measurements in anaesthetized guinea-pigs can reliably predict, during preclinical drug development, the risk of human QT prolongation. An important finding of the study was that a 5% increase in QTc (over vehicle) is a useful cutoff value for detecting significant QTc prolongation….. Compounds with hERG IC50 values above 10 μm usually do not prolong QTc in anaesthetized guinea-pigs.
S. Kongsamut et al. European Journal of Pharmacology 450 (2002) 37-41 have examined the influence of HERG affinity on QT-prolongation, and found for a series of antipsychotic drugs, the ratio of total plasma concentration to HERG IC50 ([Total plasma]/HERG IC50 )appeared to correspond well with the observed changes in QTc. This ratio ranged from approximately 11 for thioridazine (which displayed an almost 30 ms prolongation in QTc) down to 0.03 for olanzapine (1 ms prolongation in QTc).
Comparison of QTc changes, HERG IC50 and plasma concentrations for various antipsychotic drugs
| Drug | DQTc (ms) | HERG IC50 (nM) | [Total plasma](nM) | [Total plasma]/HERG IC50 |
|---|---|---|---|---|
| Thioridazine | ||||
| Ziprasidone | ||||
| Quetiapine | ||||
| Risperidone | ||||
| Olanzapine |
There is an increasing awareness that whilst HERG binding is a major contributor to QT-prolongation it far from the complete story, a number of other ion channels play important roles in the cardiac action potential and we need to understand the integrated action of drug molecules at all these channels. There are ongoing attempts to predict the cardiac effects of drugs based on their in vitro activity at a selection of ion channels, Evaluation of an in silico cardiac safety assay: Using ion channel screening data to predict QT interval changes in the rabbit ventricular wedge, Journal of Pharmacological and Toxicological Methods Volume 68, Issue 1, July–August 2013, Pages 88–96 DOI. In this study they evaluated the ability of an in silico action potential simulation assay, which uses concentration–effect data from high-throughput ion channel screens (hERG, NaV1.5, CaV1.2), to predict the results of the rabbit left-ventricular wedge assay. The highest level of predictivity for both QT prolongation and shortening is demonstrated when using PatchXpress assay data (70-80%). It should be noted that the rabbit wedge does not necessarily predict the effects in vivo, where distribution and metabolism can have an impact.
Avoiding hERG-liability in drug design via synergetic combinations of different (Q)SAR methodologies and data sources: a case study in an industrial setting. DOI
hERGcentral
A publication by Fang et al DOI describes hERGCentral: A Large Database to Store, Retrieve, and Analyze Compound-Human Ether-à-go-go Related Gene Channel Interactions to Facilitate Cardiotoxicity Assessment in Drug Development. The hERGCentral database hergcentral.org is based on experimental data obtained from a primary screen by electrophysiology against more than 300,000 structurally diverse compounds screened at 1 and 10uM. Unfortunately the database appears to be no longer available. Whilst the supplementary information for the original publication does not contain the structures of the tested compounds it does reference the PubChem substance ID. I used these identifiers to download the structures of the >300,000 records and combined them with the experimental data provided in the Excel tables in the supplementary information. The complete dataset can be downloaded here in either
or in
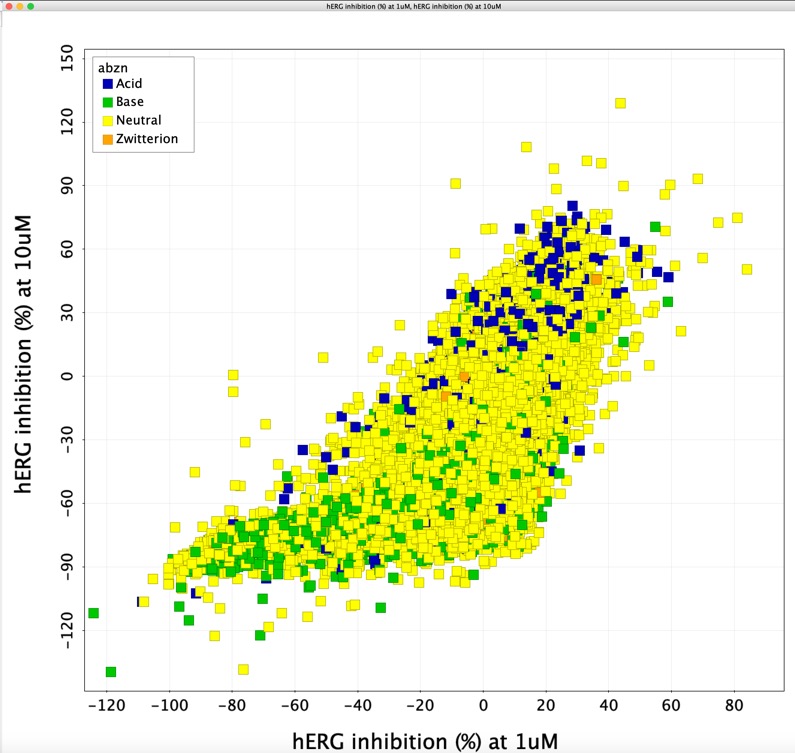
Bear in mind this is not dose response data and there will be a fair amount of scatter.
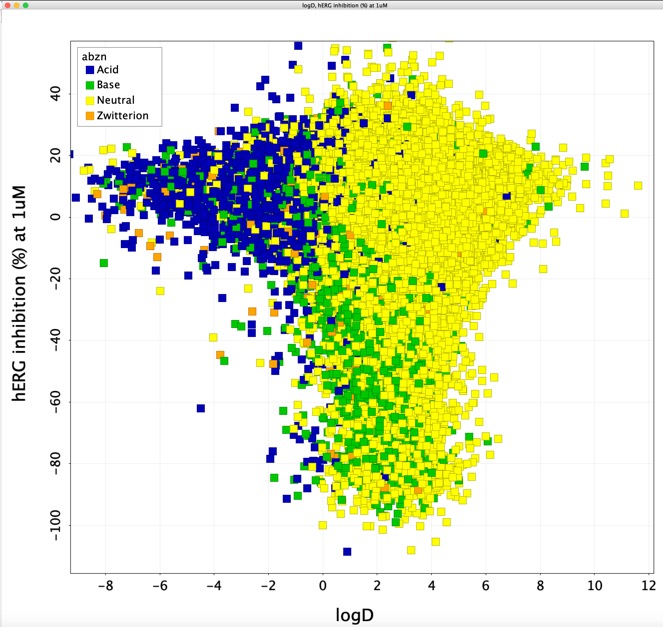
However a plot of calculated LogD (calculated using a Vortex script) versus hERG inhibition (%) at 1 uM underlines the observation that acids (blue) in general do not interact with the hERG channel and that bases (green) and neutral (yellow) are more likely to interact. In addition most of the molecules that cause hERG inhibition have a calculated LogD > 0.
Worth reading:-
The FDA Guidance for Industry, E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs
Medicinal Chemistry of hERG Optimizations: Highlights and Hang-Ups, Jamieson, DOI
Evaluation of an in silico cardiac safety assay: Using ion channel screening data to predict QT interval changes in the rabbit ventricular wedge, Journal of Pharmacological and Toxicological Methods Volume 68, Issue 1, July–August 2013, Pages 88–96 DOI.
Global Analysis Reveals Families of Chemical Motifs Enriched for hERG Inhibitors DOI.
Updated 11 February 2019