Cytochrome P450 interactions
Nearly all Drug-Drug Interactions (DDI) are due to Phase I enzymes, usually cytochrome P450 enzymes. Many drug interactions are due to the impact one drug can have on the metabolism of a second drug. One major system involved in metabolic drug interactions is the enzyme system comprising the cytochrome P450 oxidases. This system may be affected by simple saturation of the enzyme by a substrate, or either enzyme induction or enzyme inhibition or in some cases both, potentially causing significant changes in the plasma levels of administered drugs.
- Enzyme induction - Drug A induces the body to produce more of an enzyme which metabolises drug B. This reduces the effective concentration of drug B, which may lead to loss of effectiveness of drug B. Drug A effectiveness may or may not altered.
- Enzyme inhibition - Drug A inhibits the enzyme metabolising drug B, thus an elevation of drug B occurs possibly leading to an overdose.
- Bioavailability - Drug A influences the absorption of drug B perhaps by blocking active uptake mechanism or inhibiting active transport mediated excretion in the gut.
Many of the major CYP450s are now well characterised and typical substrates, inhibitors or inducers documented, there is a comprehensive list of compounds interacting with CYP450s here. Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine (2007). http://medicine.iupui.edu/clinpharm/ddis/table.asp. The CYP450 Engineering database (CYPED) provides a systematic comparison of protein sequences and structures. It was built up based on an extensible data model that enables its functions readily enhanced. The latest version of the CYPED contains information on 52,675 sequences and 595 structures respectively, which strictly follow Nelson's classification rules for homologous families and superfamilies. To gain biochemical information on substrates and inhibitors, the CYPED was linked to the Cytochrome P450 Knowledgebase (CPK). Full details are described http://www.biomedcentral.com/1471-2091/10/27.
Cytochrome P450 Inhibitors and Pharmacophores
As might be expected based on the substrate specificities there are wide range of known inhibitors and the structure-activity has been elucidated to some extent.
The X-ray crystal structures are available for some of the isoforms.
CYP 1A2
PDB Code 2HI4 Crystal Structure of Human Microsomal P450 1A2 in complex with alpha-naphthoflavone DOI.
Many inhibitors are small planer molecules, potent inhibitors include Fluvoxamine, Ciprofloxin (and other fluoroquinolone antibiotics).

I’ve added a brief analysis of the CYP2C9 inhibitor data present in ChEMBL.
CYP 2C9
PDB code 1R9O Crystal Structure of P4502C9 with Flurbiprofen bound. DOI
PDB code 1OG5 Crystal Structure of Human Cytochrome P450 2C9 with Bound Warfarin. DOI.
PDB code 4NZ2 Structure-based ligand design to overcome CYP inhibition in drug discovery projects DOI.
Potent inhibitors Fluconazole.

The x-ray structure of human P450 2C9 complexed with flurbiprofen 1R9O has been solved. Arg-108 hydrogen bonds with Asp-293 and Asn-289 on helix I and interacts directly with the carboxylate of flurbiprofen. These interactions position the substrate over the iron for regioselective oxidation in a relatively large active site cavity.

I’ve added a brief analysis of the CYP2C9 inhibitor data present in ChEMBL.
CYP 2C19
PDB code 4GQS Structural Characterization of Human Cytochrome P450 2C19: ACTIVE SITE DIFFERENCES BETWEEN P450s 2C8, 2C9, AND 2C19 DOI.
Potent inhibitors Omeprazole, moclobemide, Fluvoxamine

A series of omeprazole-based analogues have been assessed for inhibitory activity against CYP2C19 to build a pharmacophore model, Ligand-based design of a potent and selective inhibitor of cytochrome P450 2C19. DOI.
I’ve added a brief analysis of the CYP2C19 inhibitor data present in ChEMBL.
CYP 2D6
PDB code 2F9Q Crystal Structure of Human Cytochrome P450 2D6. DOI
PDB code 4WNV Contributions of Ionic Interactions and Protein Dynamics to Cytochrome P450 2D6 (CYP2D6) Substrate and Inhibitor Binding. DOI.
PDB code 5TFT Structure of cytochrome P450 2D6 (CYP2D6) BACE1 inhibitor complex DOI.
Many inhibitors are basic planer molecules, potent inhibitors include Bupropion, Fluoxetine, Paroxetine, Quinidine.

Owing to the polymorphic nature of CYP2D6, clinically significant issues can arise when drugs rely on that enzyme either for clearance, or metabolism to an active metabolite. Cytochrome P450 2D6 (CYP2D6) is absent in 7% of the Caucasian population (PM, poor metabolizers). There is also a population that has expression of multiple genes leading to the Ultrarapid Metabolizer phenotype (UM) that is present in 3% of the Caucasian population and 20–29% in Saudi Arabia/Ethiopian. CYP2D6 is involved in the metabolism of approximately 25% of drugs. The CYP2D6 inhibitor pharmacophore has been described by Strobel et al, they used the high affinity inhibitor Ajmalicine (3nM) as the template to develop the model.
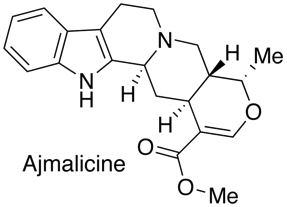
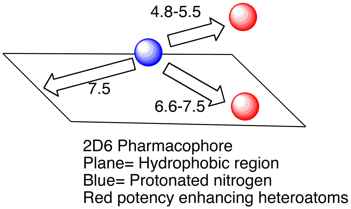
Based on molecular modeling and enzyme inhibition studies, the pharmacophore was defined by a positively charged nitrogen atom (possibly interacting with ASP301 and/or Glu216)(blue) and a flat hydrophobic region (plane) that maximally extends up to a distance of 7.5 A from this charged nitrogen atom. Compounds showing only these minimum requirements were only weak inhibitors, however. A higher inhibitory potency was observed when additional groups with a negative molecular electrostatic potential and hydrogen bond acceptor properties (red) were present at distances of 4.8-5.5 A and 6.6-7.5 A from the nitrogen atom, respectively. Whilst useful for modeling studies and offering structural insights the data set used is relatively small and probably not sufficient for general screening. More recently Ekins et al have proposed rapid computational filters based on a data set of 1750 molecules using a Random Forest classification. This model correctly identified the most potent inhibitors, however such models tend to be difficult for the bench chemist to interpret the key structural features. There is a recent review of 2D6 drug interactions which emphasizes the importance of the basic center in the ligand (In a data set of 3500 compounds of the 2D6 IC50 < 1uM 92% were basic), lipophilicity (clog P) also resulted in greater CYP2D6 inhibition for basic and neutral compounds (45% of bases with 1 < clog P < 3 exhibited 2D6 IC50 < 10 uM, in contrast 72% of bases with 3 < clog P < 5 exhibited 2D6 IC50 < 10 uM).
Strategies to reduce 2D6 inhibition
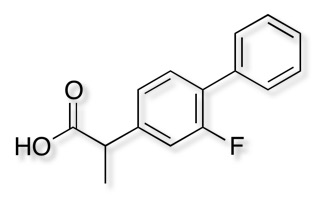
For molecules that contain a heteroatom that might coordinate to the iron (imidazoles, pyridines, triazoles, thiadiazoles etc.) substitution adjacent to the nitrogen or modifying the pKa can help. As can be seen in the example below substitution on the thiadiazole thought to bind to the iron has a dramatic effect on inhibition.
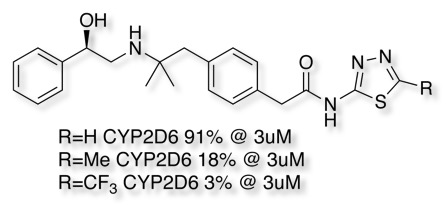
For compounds that don’t bind to the haem iron, reduction of lipophilicity (cLogP) can have a significant effect as shown in the example below.
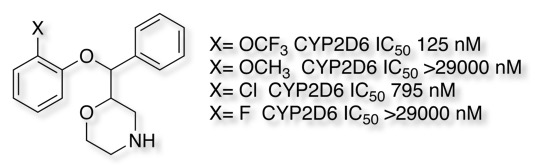
I’ve added a brief analysis of the CYP2D6 inhibitor data present in ChEMBL.
CYP 3A4
PDB code 2V0M Structural Basis for Ligand Promiscuity in Cytochrome P450 3A4 DOI.
PDB code 2J0D CRYSTAL STRUCTURE OF HUMAN P450 3A4 IN COMPLEX WITH ERYTHROMYCIN DOI.
PDB code 4K9V Dissecting Cytochrome P450 3A4-Ligand Interactions Using Ritonavir Analogues DOI.
PDB code 4D78 Structure-Based Inhibitor Design for Evaluation of a Cyp3A4 Pharmacophore Model DOI
Potent inhibitors Indinavir, clarithromycin, ketoconazole telithromycin
CYP3A4 is probably the most important enzyme in drug metabolism (J Pharm and Exp Ther 290 (1), 429, 1999). The large lipophilic binding site can accommodate a wide variety of inhibitors. Many diverse known inhibitors, often include heterocycle that binds to the haem iron in the active site, for example the anti-fungal ketoconazole shown below.

A crystal structure of Ketoconazole bound to human CYP3A4 is available, this is the crystal structure 2V0M displayed in moe, ketoconazole is shown in yellow and the haem in red. The nitrogen of the imidazole is bound to the iron, the surface of the protein is coloured coded green= lipophilic, purple= polar. The majority of the binding site is open and lipophilic and even a relatively large molecule like ketoconazole is comfortably accommodated. Other than the coordination to the iron the other major interactions are arene-arene stacking.
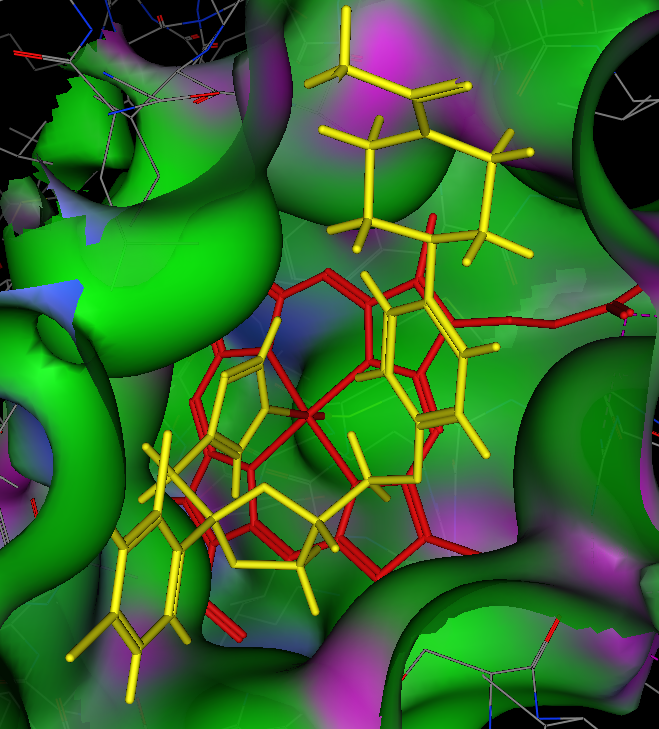
Below is the crystal structure visualised using 3Dmol.js you should be able to see the Ketoconazole imidazole (green) coordinating to the porphyrin (blue), in addition you will see that two Ketoconazole molecules are accommodated in the active site.
Mouse Controls
| Movement | Mouse Input | Touch Input | ||
|---|---|---|---|---|
| Rotation | Primary Mouse Button | Single touch | ||
| Translation | Middle Mouse Button or Ctrl+Primary | Triple touch | ||
| Zoom | Scroll Wheel or Second Mouse Button or Shift+Primary | Pinch (double touch) | ||
| Slab | Ctrl+Second | Not Available |
Strategies to reduce 3A4 inhibition
For molecules that contain a heteroatom that might coordinate to the iron (imidazoles, pyridines, triazoles) substitution adjacent to the nitrogen or modifying the pKa can help. Attempts to reduce inhibition by substitution elsewhere are often futile, the binding site is so large that the molecule simply binds in a slightly different pose. Many computational models have been built and LogD is a term that is frequently highlighted, given the lipophilic nature of the active site it is not surprising that the introduction of polar substitution is often beneficial, this may also reduce the partitioning into the liver. If arene-arene stacking is likely to be important modification of the ring electrostatics can also reduce binding.
Other resources
CYPlebrity: Machine learning models for the prediction of inhibitors of cytochrome P450 enzymes DOI, https://nerdd.univie.ac.at/cyplebrity/, structures can be submitted in SMILES format or drawn using the sketcher. The calculation takes a few second per compound and the results are displayed as shown below.
Worth reading Computational Prediction of Metabolism: Sites, Products, SAR, P450 Enzyme Dynamics, and Mechanisms J. Chem. Inf. Model. 2012, 52, 617−648.
WhichCyp: Prediction of Cytochromes P450 Inhibition, Bioinformatics, 2013, 29, 2051-2052 WhichCyp, a tool for prediction of which cytochromes P450 isoforms (among 1A2, 2C9, 2C19, 2D6 and 3A4) a given molecule is likely to inhibit. The models are built from experimental high-throughput data using support vector machines and molecular signatures.
Time dependent Inhibition
Inhibition of cytochrome P450 enzymes is one of the most common mechanisms resulting in clinically relevant drug-drug interactions. This inhibitory effect can either be a reversible or irreversible (time dependent) interaction. Time dependent inhibition (TDI) of cytochrome P450 usually involves irreversible covalent modification and is of particular concern because de novo synthesis of the enzyme is required in order to restore activity. The turnover half-lives for CYP450 enzymes are typically in the range T1/2 = 20- 100 hours Curr Drug Metab. 2008 Jun;9(5):384-94 DOI.
Examples of drugs know to act as TDI of CYP450 enzymes.
1-Aminobenzotriazole (several), Amiodarone (CYP3A), Amprenavir (CYP3A), Azamulin (CYP3A), Azithromycin (CYP3A), Bergamottin (CYP3A), Cannabidiol (CYP3A), Carbamazepine (CYP1A2), Chlorgyline (CYP1A2), Cimetidine (CYP2D6), Clarithromycin (CYP3A), Clopidogrel (CYP2B6), Delavirdine (CYP3A), Diclofenac (CYP3A), Dihydralazine (CYP1A2), 6,7-Dihydroxybergamottin (CYP3A), Diltiazem (CYP3A), Disulfiram (CYP2E1), Efavirenz (CYP2B6), EMTPP (CYP2D6), Enoxacin (CYP1A2), Erythromycin (CYP3A), Ethinyl estradiol (CYP3A), 2-Ethynylnaphthalene (CYP1A), Fluoxetine (CYP3A), Furafylline (CYP1A2), Gemfibrozil glucuronide (CYP2C8), Gestodene (CYP3A), Glabridin (CYP2B6), Hydrastine (several), 4-Ipomeanol (CYP3A), Irinotecan (CYP3A), Isoniazid (several), Lopinavir (CYP3A), Menthofuran (CYP2A6), Methoxsalen (CYP2A6), 3,4-Methylenedioxymethamphetamine (CYP2D6), 3-Methylindole (CYP2F), Mibefradil (CYP3A), Midazolam (CYP3A), Mifepristone (CYP3A), Nefazodone (CYP3A), Nelfinavir (CYP3A), Nicardipine (CYP3A), Paroxetine (CYP2D6), Phencyclidine (CYP2B6), Phenelzine (several), Pioglitazone (CYP3A), n-Propylxanthate (CYP2B6), Raloxifene (CYP3A), Resveratrol (CYP3A), Rhapontigenin (CYP1A1), Ritonavir (CYP3A), Rofecoxib (CYP1A2), Rosiglitazone (CYP3A), Roxithromycin (CYP3A), Rutaecarpine (CYP1A1, 1B1), Saquinavir (CYP3A), Silybin (CYP3A), Suprofen (CYP2C9), Tabimorelin (CYP3A), Tamoxifen (CYP3A), ThioTEPA (CYP2B6), Ticlopidine (CYP2B6, 2C19), Tienilic acid (CYP2C9), Troglitazone (CYP3A, 2C8, 2C9), Troleandomycin (CYP3A), Verapamil (CYP3A), Zafirlukast (CYP3A), Zileuton (CYP1A2).
The biochemical mechanisms by which compounds function as mechanism-based P450 inactivators can be divided into three categories: quasi-irreversible or metabolite-intermediate complex formation, heme alkylation, and protein alkylation.
| Substituent | Possible Reactive intermediate | Example |
|---|---|---|
| Aliphatic amine | Nitroso | Clarithromycin |
| Alicyclic amine | Iminium ion | Phencyclidine |
| Cyclopropylamine | Radical | Tranylcypromine |
| Methylenedioxyphenyl | Carbene | Paroxetine |
| Furan | α,β-Unsaturated carbonyl or epoxide | Methoxsalen |
| Thiophene | S-oxide or epoxide | Tienilic Acid |
| Alkenes | Cation radical | Secobarbital |
| Alkynes | Ketene or oxirene | Gestodene |
| 2-Alkylimidazole | Imidazomethide | Furafylline |
| 3-Alkylindole | α,β-Unsaturated imine | Zafirlukast |
| Dihaloalkane | Acylhalide | Chloramphenicol |
| Hydrazine | Radical | Dihydralazine |
| Aminophenol | Quinoneimine | Nefazodone |
| Phenol | Quinone | Raloxifene |
As can be seen a number of well tolerated drugs display TDI in in vitro systems, putting the potential risk in perspective and the models are described in detail in "The conduct of in vitro studies to address time-dependent inhibition of drug-metabolizing enzymes: a perspective of the pharmaceutical research and manufacturers of America." Drug Metab Dispos. 2009 Jul;37(7):1355-70 DOI.
Worth reading "Prediction of Drug-Drug Interactions Arising From Mechanism-Based Inactivation: Key Input Parameters and Impact on Risk Assessment" DOI.
FDA DRAFT Guidance for Industry – Drug Interaction Studies (Feb. 2012)
“TDI should be studied in standard in vitro screening protocols by pre-incubating the drug... before the addition of a substrate. Any time dependent loss of initial product formation rate may indicate time dependent inhibition, and definitive in vitro studies to obtain TDI parameters (i.e., kinact and KI). Details of this tiered approach were proposed by the PhRMA Drug Metabolism Technical Group (Grimm et al. 2009).” See also idiosyncratic toxicity
Cytochrome P450 Induction
The primary mechanism of cytochrome P450 induction is via increased gene transcription which typically occurs through nuclear receptor activation. Whilst the first evidence of enzyme induction may occur after multiple doses in vivo (AUC and T1/2 reduction) there are a number of in vitro systems that can be used.
Whilst cytochrome P450 induction can be assayed in hepatocytes it is more usual to use nuclear receptor transactivation assays to assess the potential of test compounds to cause enzyme induction. PXR and AhR nuclear receptor activation can be assayed using a stably-transfected human hepatoma cell lines and a luciferase reporter gene assay in 96-well format.
Pregnane X receptor (PXR)
The pregnane X receptor (hPXR) is the major determinant of CYP3A gene regulation by drugs and other xenobiotics. In addition, PXR mediates induction of P450s 2B6, 2C8/9, and 3A4, as well as the drug transporters MDR1, organic anion transporting polypeptide C, bile salt export protein, and multidrug resistance-associated protein 2.
Known PXR activators include

Binding site is large and hydrophobic with several important hydrogen bonding interactions. May be multiple binding conformations. Similar to CYP3A pharmacophore, many (but not all) CYP3A substrates/inhibitors are also CYP3A inducers. There have been a number of in silico models built to predict PXR activation Jacobs et al used PLS and VolSurf descriptors for the development of a QSAR model for PXR/AhR interaction DOI. Xiao et al used docking into a modeled PXR ligand-binding domain that takes into consideration the structural variations of five published X-ray structures of PXR–ligand complexes DOI.
Low molecular weight (MW < 300) compounds were in general found to be non-binders, and those molecules that do not match the shape of the PXR ligand-binding site may also act as a non-binder. Secondly, the favorable hydrophobic interactions, mostly through aromatic π–π interactions, and the presence of suitable hydrogen bond(s) between the compounds and PXR are attributes of strong binders.
Machine learning techniques have also been applied (support vector machine (SVM), k-nearest neighbor (k-NN), and artificial neural networks (ANN)) DOI using 73 descriptors selected from a pool of over 500 descriptors, whilst this method has the advantage of speed and capacity, it is much more difficult for the medicinal chemist to interpret and decide what to make next.
In an in silico study looking at currently prescribed drugs a combination of machine learning and docking studies were able to identify previously unknown PXR activators, interestingly several compounds were poorly predicted by the docking studies. Perhaps because as the authors noted
In general, we observed that because of the relatively large size of the binding pocket, compounds may move around and interact with different sites within the binding pocket.
The PDB structure of PNU-142721 bound to PXR highlights a number of the issues with docking studies, PDB 3R8D
PXR employs one hydrogen bond and fourteen van der Waals contacts to interact with the ligand, but allows two loops adjacent to the ligand-binding pocket to remain disordered in the structure. These observations highlight the role structural flexibility plays in PXR's promiscuous responses to xenobiotics.


PNU-142721 bound into the active site of PXR (Green surface=hydrophobic, Purple=H-bond, Blue =Polar)
Aryl hydrocarbon receptor (AhR)
The aryl hydrocarbon receptor (AhR) is a member of the basic helix–loop–helix (bHLH)-Per-ARNT-Sim (PAS) family of transcriptional regulators that control a variety of developmental and physiological events. Known ligands include ligands such as TCDD, coplanar polychlorinated biphenyls (PCBs) and dibenzo[a]antracene (DBMA). Ligand binding to the AhR is presumed to produce conformational changes in the AhR protein which result in the exposure of an AhR nuclear localization signal and the translocation of the whole complex into the nucleus where at the activated transcription factor complex is formed. This then binds to specific sequences of DNA resulting in increased transcription. Whilst CYP1A1 was the first cytochrome shown to be induced by TCDD an “Ahr gene battery” of Phase I and Phase II metabolizing enzymes consisting of CYP1A1, CYP1A2, CYP1B1, NQO1, ALDH3A1, UGT1A2 and GSTA1 have subsequently been identified.

Tobacco smoking has also been shown to induce CYP1A via activation of AhR. AhR also interacts with other signalling pathways such as those mediated by Estrogen Receptor and other Hormone receptors, Hypoxia, NF-KappaB and Rb and it appears to be directly involved in the reproductive toxicity caused by a range of aryl hydrocarbons, including the highly toxic compound 2,3,7,8-tetrachlor-odibenzo-p-dioxin (TCDD), DOI : 10.1530/rep.1.00294
In silico models have been developed Jacobs et al used PLS and VolSurf descriptors for the development of a QSAR model for PXR/AhR interaction DOI. Modeling of the Aryl Hydrocarbon Receptor (AhR) ligand binding domain and its utility in virtual ligand screening to predict new AhR ligands has been described DOI, this work also compared different species.
Constitutive androstane receptor (CAR)
The xenobiotic receptor constitutive androstane receptor (CAR) mediates the well-studied induction of CYP2B genes and other drug-metabolizing enzymes by phenobarbital (PB), an antiepileptic drug that has been shown to alter thyroid hormone (TH) levels. Unlike most nuclear receptors, this transcriptional regulator is constitutively active in the absence of ligand but is regulated by both agonists and inverse agonists. Ligand binding results in translocation of this protein to the nucleus, where it activates or represses target gene transcription.

Efavirenz is metabolised in the liver, and is both a substrate and inducer of the 2B6 and 3A4 isoforms of the cytochrome P450 system.
a 3D-QSAR model has been developed DOI using a combination of protein structure and ligand based methods.
Updated 5 September 2021