Directed and Virtual Screening
Whilst high-throughput screening (HTS) has been the starting point for many successful drug discovery programs the cost of screening, the accessibility of a large diverse sample collection, or throughput of the primary assay may preclude HTS as a starting point and identification of a smaller selection of compounds with a higher probability of being a hit may be desired. Directed or Virtual screening is a computational technique used in drug discovery research designed to identify potential hits for evaluation in primary assays. It involves the rapid in silico assessment of large libraries of chemical structures in order to identify those structures that most likely to be active against a drug target. Whilst there are a wide range of techniques available they fall into two main categories, 1) Ligand-based and 2) Structure-based.
Ligand-Based
Given one or more ligands that bind to the target, these are described using simple descriptors which are then used to rapidly search chemical databases. The descriptors used can be simple functional groups, atom pairs or pharmacophores. Ligand-based methods typically require a fraction of a second for a single structure comparison operation.
Structure-based
Given the structure of the target protein potential ligands are evaluated for their ability to bind to the active site, usually this is a much more lengthly process than ligand-based screening. The process is usually thought of in two components docking and then scoring.
Ligand-based Methods
Substructure searching
Given a known ligand or group of ligands it may be possible to identify a key structural feature that is important for binding, a search for this substructure in a chemical database would then be used to identify compounds for screening. This technique is designed to identify exact copies of the query feature within the molecular structure.
Molecular similarity methods
In similarity searching, an abstract molecular representation in descriptor space is calculated which is compared to abstract representations of other molecules, the difficulty is knowing the best descriptors to use, there potentially an infinite number of descriptors and the challenge is to select the most appropriate, bearing in mind the time taken to calculate a descriptor and the potentail overlap with other descriptors. Todeschini and Consonni defined a molecular descriptor as:
“The molecular descriptor is the final result of a logic and mathematical procedure which transforms chemical information encoded within a symbolic representation of a molecule into a useful number or the result of some standardized experiment”
The descriptor can be a physicochemical property such as LogP, pKa or a theoretical molecular representation of the molecule such as atom counts or atom pair paths. The simplest descriptors are counts of individual atoms, bonds, rings, hydrogen bond donor/acceptor, acid/base.
2D descriptors can encode further information about the molecule these include augmented atoms, atom sequences, atom pairs topological torsions and ring types.
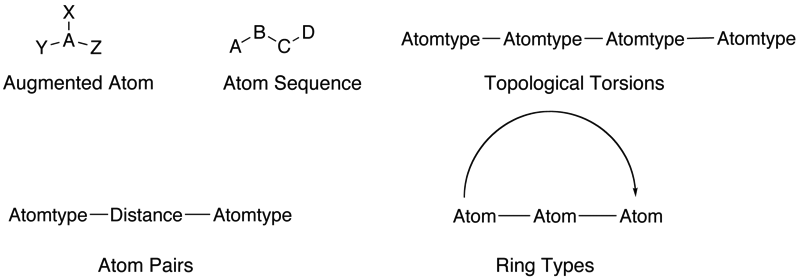
3D descriptors include atom triangles, distance and angle based descriptors, these will require a 3D structure of the molecule and will depend on conformation.
View a list of descriptors
Shape-Based Methods
OpenEye have developed ROCS a fast molecular shape comparison tool, it uses a smooth Gaussian function to represent the molecular volume, so it is possible to routinely minimize to the best global match. Although ROCS is primarily a shape-based method, a number of functions can be used to "colour" the forcefield to improve matching and post-shape scoring.
Donor: Functional groups that can act as H-bond donors e.g. acid-OH Acceptor: Functional groups that can act as H-bond acceptors e.g. carboxylate Anion: Functional groups with either localized or delocalized negative charge e.g. tetrazole Cation: Functional groups with either localized or delocalized positive charge e.g. guanidinium Hydrophobe: Terminal or non-terminal aromatic or aliphatic groups e.g. phenyl Rings: Rings of defined size e.g. 4-7 atoms
I’ve included a list of descriptors here.
Fingerprints
A structural fingerprint is a bitstring in which each bit represents the presence (TRUE) or absence (FALSE) of a specific structural feature or descriptor. For example the MACCS Structural fingerprint indicates the presence of each of the 166 public MDL MACCS structural keys computed from the molecular graph. The fingerprint is represented as a sparse list of keys present in the molecule. Structural keys vary widely in size, from a few tens or hundreds of bits to several thousand bits. The choice of size is a tradeoff between specificity and space. However Structural fingerprints are usually very "sparse" (mostly zeros) since a typical molecule has very few of the patterns that the structural key's bits represent.
The other major fingerprint type is called "hash fingerprints." The most well known of these are the Daylight fingerprints. These enumerate all linear substructures of length N in the molecule. Daylight sets the bounds on N as 3 <= N <= 7. The list of patterns produced is exhaustive: Every pattern in the molecule, up to the pathlength limit, is generated. Hash fingerprints are much denser, and because there are no predefined structural features they are able to accommodate all types of molecules.
MolPrint2D is an atom-environment fingerprint developed by Bender et al which has been used in QSAR studies and for measuring molecular similarity. Andreas Bender, Hamse Y. Mussa, and Robert C. Glen. Molecular Similarity Searching Using Atom Environments, Information-Based Feature Selection, and a Naive Bayesian Classifier. J. Chem. Inf. Comput. Sci. 2004, 44, 170-178.
Multilevel Neighborhoods of Atoms (MNA) descriptors are 2D molecular fragments suitable for use in QSAR modelling. The format outputs a complete descriptor fingerprint per molecule. Thus, a 27-atom (including hydrogen) molecule would result in 27 descriptors, one per line. MNA descriptors are generated recursively. Starting at the origin, each atom is appended to the descriptor immediately followed by a parenthesized list of its neighbours. This process iterates until the specified distance from the origin, also known as the depth of the descriptor. Dmitrii Filimonov, Vladimir Poroikov, Yulia Borodina, and Tatyana Gloriozova. Chemical Similarity Assessment through Multilevel Neighborhoods of Atoms: Definition and Comparison with the Other Descriptors. J. Chem. Inf. Comput. Sci. 1999, 39, 666-670.
Spectrophores are one-dimensional descriptors generated from the property fields surrounding the molecules. This technology allows the accurate description of molecules in terms of their surface properties or fields. Comparison of molecules’ property fields provides a robust structure-independent method of aligning actives from different chemical classes. When applied to molecules such as ligands and drugs, Spectrophores can be used as powerful molecular descriptors in the fields of chemoinformatics, virtual screening, and QSAR modeling. The calculation of a Spectrophore consists of determining the total interaction value V(c,p) between each of the atomic contributions of property p with a set of interaction points on an artificial cage c surrounding the molecular conformation.
Fingerprints in Openbabel
At present there are four types of fingerprints: FP2, a path-based fingerprint which indexes small molecule fragments (somewhat similar to the Daylight fingerprints), fingerprint types FP3 and FP4 which both use a series of SMARTS queries that are stored in patterns.txt and SMARTS_InteLigand.txt, and a MACCS fingerprint that uses the SMARTS patterns in MACCS.txt.
Similarity
Whilst finding an exact match is usually easy, finding a “similar” compound requires us to agree on how we describe a molecule and there are huge number of potential list of descriptors.

Which is the most similar to the Fire Engine?



The ambulance because it is an emergency vehicle, the red barn because of the colour, or the glass because it contains water?
The most widely used and accepted fingerprint similarity function in chemical informatics is called the Tanimoto coefficient.
c/(a+b+c)
Where:
a is the count of bits on in object A but not in object B.
b is the count of bits on in object B but not in object A.
c is the count of the bits on in both object A and object B.
d is the count of the bits off in both object A and object B.
Identical fingerprints have a score of 1.0, and when there are no bits in common the score is 0.0. From a chemists viewpoint if two molecules have a Tanimoto coefficient of > 0.7 they are recognisably similar. The most common useful similarity indexes have been collected by Holliday et al (Holliday, JD., Hu, C-Y. and Willett, P. (2002) Combinatorial Chemistry and High Throughput Screening 5, 155-166)
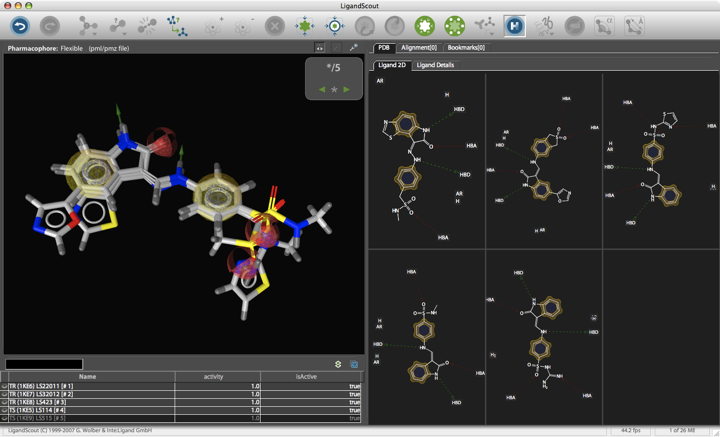
Pharmacophore-based searches
A pharmacophore is a collection of steric and electrostatic features that are necessary to ensure the interaction of a molecule with a specific biological target protein and to elicit or inhibit a biological activity. A pharmacophore does not represent an actual molecule or any specific functional group, but it accounts for the common molecular interaction capacities of a group of compounds towards their target structure. Typical features used in pharmacophore-based searches include hydrogen bond acceptor, hydrogen bond donor, charge positive, charge negative, lipophilic, aromatic and may be described by ring centers or virtual points in space.
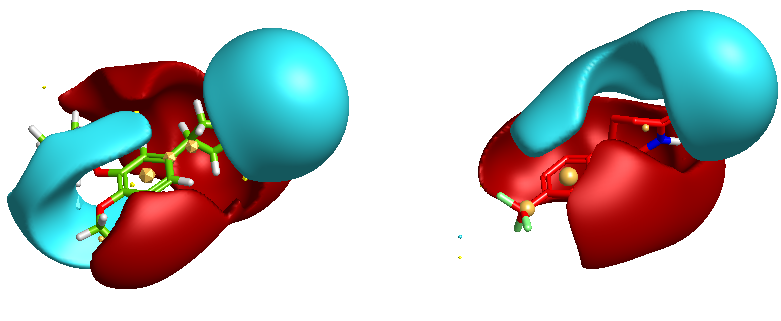
Field-Based Methods
Cresset have pioneered the use of field point descriptors to give a more accurate description of the charge around an atom and to enable better comparisons and visualization. These field points can be used to compare molecular similarity. FieldScreen compares the Fields surrounding your lead in a given 3D conformation against a database of Field pharmacophores created drug-like compounds.
Structure-based Methods
Docking is a method which attempts to predict the preferred orientation of a molecule when bound to a protein to form a stable complex. Knowledge of the preferred pose in turn may be used to predict the strength of association or binding affinity between two molecules using scoring functions.
This requies a database of molecules, some docking programs generate conformations on the fly others require prior generation of multiple potential low energy conformations either using systematic generation, or a stochastic sampling of conformations. It is important to ensure that the molecules have the appropriate ionisation at ph 7.4. The quality of the protein structure is critical, the higher the resolution the better, in particular those atoms in the binding site (Check B-factors, high numbers suggest protein mobility). Since conformational changes can take place upon ligand binding having a structure with ligand bound is extremely useful.
Docking Algorithms
The principal docking methods that are used extensively employ search algorithms based on Monte Carlo, genetic algorithm, fragment-based and molecular dynamics, some examples are shown in the list below.
AutoDock http://autodock.scripps.edu/
AutoDock Vina is reported to be orders of magnitude faster than AutoDock whilst improving binding mode predictions.
FRED http://www.eyesopen.com/products/applications/fred.html
Smina is a fork of Autodock Vina that focuses on improving scoring and minimization. More details are disclosed in this publication DOI.
Scoring function
The scoring function takes a pose as input and returns a number indicating the likelihood that the pose represents a favorable binding interaction. Most scoring functions are physics-based molecular mechanics force fields that estimate the energy of the pose; a low (negative) energy indicates a stable system and thus a likely binding interaction. An alternative approach is to derive a statistical potential for interactions from a large database of protein-ligand complexes, such as the Protein Data Bank, and evaluate the fit of the pose according to this inferred potential.
Force-field scoring functions. Make use of classical molecular mechanics for energy function calculations. The binding free energy of protein-ligand complexes are estimated by the sum of van der Waals (by Lennard-Jones potential function) and electrostatics interactions. Solvation is considered as a distance-dependent dielectric function. Non- polar contributions are assumed to be proportional to the solvent-accessible surface area. Non-bonded interactions are treated with the introduction of a cut-off distance. The method requires energy minimization of the complex prior to energy evaluations. Various force-field scoring functions are based on different sets of parameters.
Empirical scoring functions. The binding free energy is estimated based on weighted structural parameters by fitting the scoring functions to experimental determined binding constants of a set of complexes. These scoring functions may be potentially biased by the selected training set of ligand- protein complexes. The advantage of these functions is that their terms, although similar to force-field functions, are orders of magnitude easier to evaluate. More complex functions attempt to addresses solvation and desolvation effects but since these effects are poorly understood they provide only incomplete descriptions of these effects on protein-ligand binding.
Knowledge-based scoring functions. Here, binding affinity is considered as a sum of the ligand and protein atoms interactions. As the empirical scoring functions, these potentials are derived from experimental structures, where interatomic distances are converted into distance-dependent interactions free energies. These functions are designed to reproduce experimental structures rather than binding energies. Due to its simplicity, knowledge-based scoring functions permit rapid screening of large compound databases.
Recently, machine-learning scoring functions trained on protein-ligand complexes have shown significant promise an example being (RF-Score-VS) trained on 15 426 active and 893 897 inactive molecules docked to a set of 102 targets DOI.
Our results show RF-Score-VS can substantially improve virtual screening performance: RF-Score-VS top 1% provides 55.6% hit rate, whereas that of Vina only 16.2% (for smaller percent the difference is even more encouraging: RF-Score-VS top 0.1% achieves 88.6% hit rate for 27.5% using Vina). In addition, RF-Score-VS provides much better prediction of measured binding affinity than Vina (Pearson correlation of 0.56 and −0.18, respectively). Lastly, we test RF-Score-VS on an independent test set from the DEKOIS benchmark and observed comparable results.
Virtual Screening/Docking requires a set of different tools, ligand set selection, target protein QC, conformer generation, docking, scoring, pose selection, vendor selections and ordering.
Workflows describing the process have been published, A workflow for docking/virtual screening.
Updated 8 April 2024