
Much of medicinal chemistry is based on the optimisation or reduction of interactions between a small molecule and a variety of biomolecules, this can be increasing the affinity of a ligand for a receptor or reducing affinity for some undesired off-target interaction such as HERG or CYP450. Whilst the overall physicochemical properties of the molecule can have a major influence it likely that specificity might be driven by optimisation of strength and geometry of specific molecular interactions.
Strength of interactions
Whilst the strength of a covalent single bond is usually in the region 80-100 Kcal/mol the non-covalent interactions exploited by medicinal chemists are much weaker. Andrews has tried to estimate the average strength of various molecular interactions by examining the structural components and binding affinities of 200 compounds. Other have tried to estimate the strength of interaction by using chemical double mutants.
Typical Energies
Salt Bridge ~2 kcal/mol
H-Bond ~1 kcal/mol
Hydrophobic ~0.7 kcal/mol
Aromatic ~1-3 kcal/mol
Since dG=-2.303RTlogK we can calculate that a single ionic interaction might afford a 25-fold increase in affinity, whilst a hydrogen bond yield a 6-fold increase, 3.5-fold increase in binding constant for a methyl group. However it is important to note that steric clashes can have a much more pronounced impact on affinity, the interaction between two atoms is described by the Lennard-Jones potential shown graphically below.
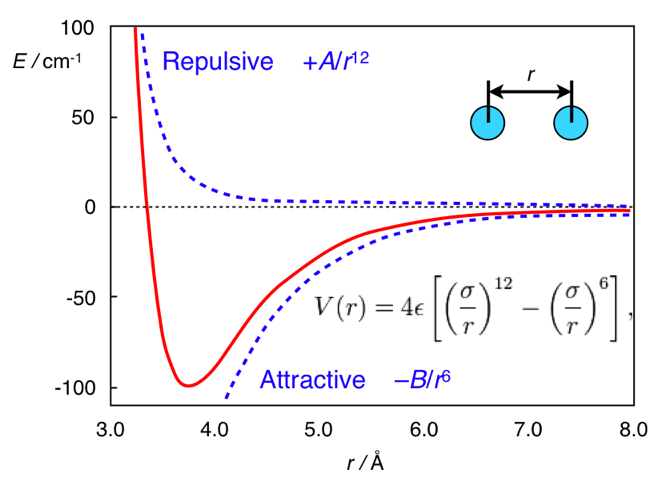
As you can see the attractive forces predominate when the atoms are further apart but when they get too close the repulsive forces become dramatically dominant. Hence a small steric clash can cause the loss of all affinity.
Specific Interactions
With many of these interactions simply looking at a model of a protein with a ligand docked can be misleading, only considering the bound state only gives part of the equation. Desolvation plays a very important role, particularly with ionised or polar functional groups where there will be a large unfavourable desolvation term, in addition entropic changes can also have a significant influence.
Salt Bridges
For many biogenic amines a key requirement for ligand recognition is the interaction between a protonated amine in the ligand and a specific aspartic acid residue buried in the membrane domain of a GPCR. Kumar has shown that the 3D structure of many proteins is stabilised by internal salt bridges, many buried within the core of the protein. In many cases a salt bridge is really a combination of hydrogen bonding and an ionic interaction. Estimates for the strength of such interactions can be misleading since there is a large unfavourable desolvation term and for mobile residues on the surface of proteins there may be a significant entropic loss. However in the case of salt bridges buried within the core of the protein there are many examples where they have been shown to be highly stabilising. One of the most stabilising salt bridges is formed by residues Glu27 and Arg387 in the human salivary a-amylase (PDB code: 1smd), shown in the image below. Whilst there is a significant desolvation penalty, the side chain centroids are 3.7 Angstroms apart, and the measured hydrogen bond length is only 1.94 Angstroms. The side-chain ionised carboxylic acid oxygen atoms of Glu27 form two very strong hydrogen bonds with the side-chain protonated nitrogen atoms in the guanidinium group of Arg387, with the additional possibility of extra weaker cross h-bonds increasing stability. dG for this salt bridge has been calculated to be -22.4 kcal/mol.
Hydrogen Bonds
The Hydrogen bond is a ubiquitous element of the recognition in biological systems. Estimation of the energetic contribution from hydrogen bonds is sometimes problematic, since in aqueous solution formation of a hydrogen bond requires the desolvation of both donor and acceptor. However current estimates range from 1-40 kcal/mol. The higher energy hydrogen bonds occur in the case of charge reinforced hydrogen bonds (see above). The length of the Hydrogen bond ranges from 2.6 Angstroms to 3.1 Angstroms based on observation from the PDB. (Structure in Protein Chemistry, Jack Kyte
| Glu27 and Arg387 in the human salivary a-amylase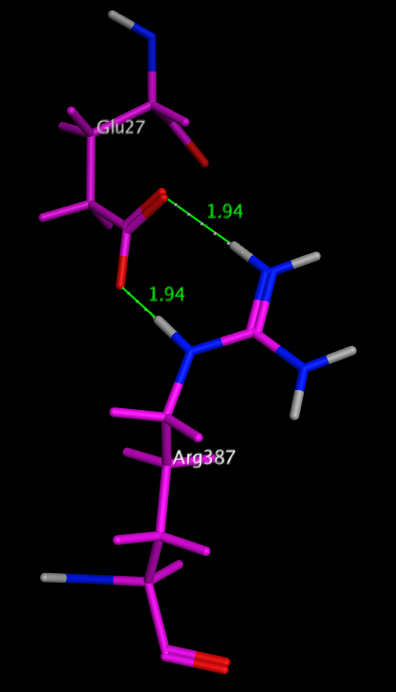 |
|---|
The geometry of these interactions has been studied and the preferred geometries identified for a number of functional groups (Journal of Computer-Aided Molecular Design, 10 (1996) 607-622) based on data taken from CSD.
Hydrogen-bonding properties have often been correlated with pKa data, and within a chemical series e.g substituted pyridines this may well be true but this is not true when comparing unrelated functional groups, For example, thiols are much more acidic than alcohols, because they can stabilize the charged anionic state formed on deprotonation, but they are much worse hydrogen-bond donors, because they are less polar.
Intramolecular hydrogen bonding can be important for stabilizing a particular ligand conformation, Stahl et al (J. Med. Chem. 2010, 53, 2601–2611) DOI: 10.1021/jm100087s , have completed an extensive search of the CSD and shown the highest frequency of
intramolecular hydrogen bonds have planar, six-membered rings stabilized by conjugation with a π-system. A variety of further, far less explored topologies have been identified: Weaker six-membered ring hydrogen bonds containing one sp3 center and, in particular, a number of nonplanar seven-membered and eight-membered ring topologies. Five-membered ring intramolecular hydrogen bonds have the smallest angles and the longest H-bond distances. With N-H as the proton donor, C=O or heterocyclic N appear to be the preferred acceptors.
An example is shown below. 1FN
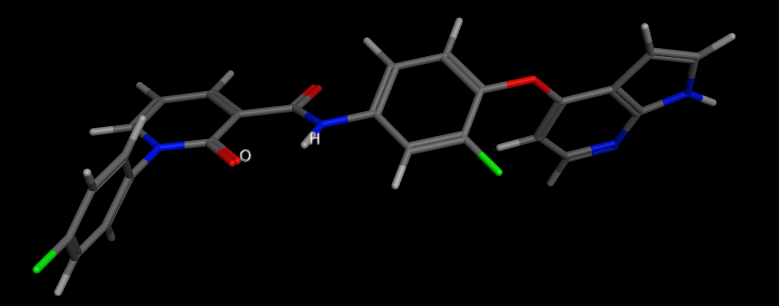
Whilst much of the crystallographic work has concentrated on the influence that hydrogen bonding has on the structure of proteins a recent paper by Selwood et al has presented an analysis of the chemical fragments that hydrogen bond to Asp, Glu, Arg and His side chains in proteins.
As one might expect the fragments found to form two hydrogen bonds to the carboxylic acid of Asp and Glu include arylamidines, guanidines and 2-amino azoheterocycles such as 2-aminopyridine. Interestingly the non-basic 1,2-cyclic diols are also common double hydrogen bond forming fragments. A wide range of fragments can form a single hydrogen bond with Asp or Glu including amines, heterocycles, sulphonamides, hydroxyls and carboxylic acids.
Arg has a pKa of 12 and is positively charged at physiological pH, therefore, Arg is entirely a hydrogen bond donor in protein ligand interactions. The most common functional groups to interact with Arg have phosphates, phosphonate, and carboxylic acids. The vast majority of the other ligands bond via an O-mediated hydrogen bond, this includes sulphonyls, ketones, phenols and ketones.
In contrast to the other side chains His with a pKa of 6 can interact in either protonated or unprotonated form. It can coordinate with many metals. Whilst acidic groups again form a proportion of the observed ligands azaheterocycles, sulphonamides, hydroxyls, and carbonyls were also observed. Full details of the fragments are available in the supporting information.
Aryl-Aryl Interactions
Interactions between aromatics rings are well documented, 60% of the aromatic residues are involved in aryl-aryl interactions. The T-shaped edge-to-face and the parallel-displaced stacking arrangement predominate. Whilst in proteins the parallel-displaced stacking arrangement is observed more often the two arrangements are of similar energy -1.6 to -2.4 kcal/mol, and is thought to be a combination of VDW dispersion and electrostatics (A word of caution, the precise nature of the interaction is poorly understood and may not be modeled accurately by molecular modeling packages). The strength of the interaction can be influenced by substitution, Stacking arrangements of an electron-poor and an electron-rich aromatic ring profit from charge transfer. Stacking between electron-deficient rings is generally preferred over stacking of electron-rich ones. In our work on NK1 antagonists the X-ray structures (shown below) illustrate how the differences in the pi-stacking interactions depend on the nature of the aromatic rings.
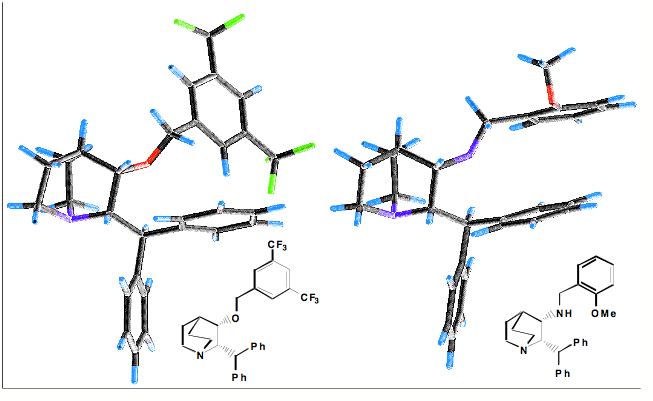
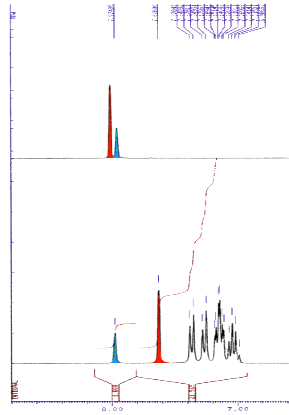
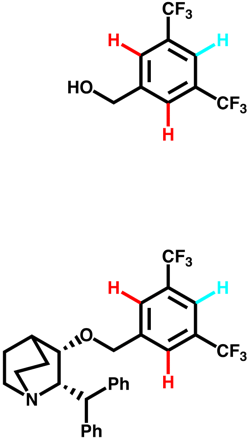
The influence of the edge to face pi-stacking interaction can be seen in the NMR, comparing the parent benzyl alcohol with the benzyl ether (below). It is apparent that in the benzyl ether the two ortho protons (red) are shifted upfield due to the shielding effect of one of the aromatic rings of the benzhydryl group.
Cation-Aryl Interactions
Dougherty, (JACS, 2000, 122, 870) Has undertaken a high level theoretical comparison of Cation-Aryl with salt bridges in a range of solvents and found the cation-aryl interaction is strong across a range of solvents including polar media, which is not found salt bridges. Part of the reason for the difference is that ionised groups must be desolvated in aqueous solution whereas aromatic rings are already desolvated to a large extent. Many cation-aryl interactions occur on the surface of proteins, and as a general rule Arg interacts more often than Lys, with Trp providing the aromatic interaction more often than Phe or Tyr.
Methyl groups interact with the face of aromatic rings when bound to an electronegative atom. A positively charged nitrogen is a particularly strong electronegative substituent, and therefore, the direct interaction of an alkylated ammonium group with an aromatic ring leads to a strongly attractive interaction at a distance of 3.4-4.0 Angstroms. It might be expected that in acetylcholine esterase (ACHE) the binding of acetylcholine could be stabilised by an interaction between the quaternary ammonium and a carboxylate of Asp or Glu, in fact it is stabilised by an interaction with a tryptophan (below).
X-ray structure of Acetylcholine esterase showing quaternary ammonium moiety of acetyl choline (yellow) stabilised by cation-aryl interaction with a tryptophan (purple).

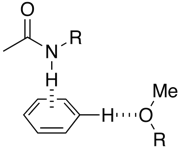
In the case of protonated amines a similar interaction is possible, but in addition the hydrogen on the nitrogen often provides additional stabilisation via a hydrogen bonding interaction.
|
 |
Worth reading:- Interactions with Aromatic Rings in Chemical and Biological Recognition, Angew. Chem. Int. Ed. 2003, 42, No. 11
Bonding Sulphur and Oxygen
Nonbonded interactions between sulphur atom and carbonyl oxygen atom have been observed in proteins and small molecules.
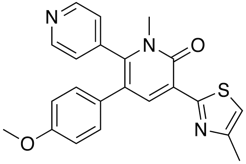
In the GABA ligand below (J. Med. Chem. 2002, 45, 1887-1900 ), whilst the he C-5 and C-6 aromatic rings (interplanar torsions ca. 58 and 63°, respectively) were twisted out of plane with the central pyridone, the thiazole was clearly coplanar with the pyridone ring (interplanar torsion ca. 3°), with a close intramolecular contact of the sulphur and the carbonyl oxygen (S...O ) 2.76 Angstrom; (van der Waals radii S 1.80 Angstrom, O 1.52 Angstrom).
Bonding to fluorine and chlorine
Bonds to halogen are significantly weaker than hydrogen bonds but there are many examples in the PDB of carbonyls interacting with halogens with bonds to I, and Br predominating. It is perhaps worth noting that halogens have been introduced into ligands to aid X-ray analysis, but they may also influence binding. Based on the observed bond angles the interaction is between the halogen and pi-cloud of the carbonyl rather than the lone pair with a clear clustering of X--O=C-N dihedral angles of 90° associated with interactions that involve primarily the pi-system of the carbonyl.
Example ligand binding interactions
A recent publication from Mileni et al (DOI: 10.1021/jm9012196) rather nicely illustrates a whole range of molecular interactions and I’ve taken the pdb file (3K83) into MOE and highlighted some of the key interactions below. The catalytic mechanism involves nucleophilic attack of Ser241 onto the amide carbonyl of the substrate, in the case of the inhibitor below (Ki 22 nM), Ser241(yellow) forms a hemiacetal with carbonyl adjacent to the oxazole. This tetrahedral intermediate is stabilised by the electron-withdrawing heterocycle and hydrogen bonds to backbone N-Hs.

Interestingly, the carboxylic acid of the ligand does not interact with a basic amine but is instead stabilised by interaction with three water molecules and hydrogen bonds to two backbone N-H. The remaining key interactions are a series of Aryl interactions. Phe192 forms an edge to face pi-interaction with the pyridyl ring of the inhibitor and with one of the rings of the biphenyl group. Phe381 makes a key
aryl CH-pi contact with the inhibitor biphenyl ring whilst two Met236 and Met495 orient their sulfur lone pair electrons toward the bound phenyl hydrogens and the hydroxyl of Thr377 also interacts with the phenyl hydrogens.
X-ray Crystallographic Analysis of alpha-Ketoheterocycle Inhibitors Bound to a Humanized Variant of Fatty Acid Amide Hydrolase
Worth reading:-
Medicinal Chemist’s Guide to Molecular Interactions. Bissantz, Kuhn, Stalh J Med Chem http://pubs.acs.org/doi/abs/10.1021/jm100112j
Quantifying Intermolecular Interactions : Guidelines for the Molecular Recognition Toolbox. Angew. Chem. Int. Ed. 2004, 43, 5310 – 5324, DOI:10.1002/anie.200301739
Hydrophobic Binding
Singh et al (American Journal of Immunology 4 (3): 33-42, 2008) have completed an extensive study on the binding interactions that are important for the binding of Tetrahydroimidazobenzodiazepinone (TIBO) which belongs to non-nucleoside group of reverse transcriptase inhibitors (NNRTIs). The study showed that hydrophobic interaction is predominant and made major contribution, while hydrogen bonding and polar interactions help in proper orientation of the compound,
The "hydrophobic effect" (a term coined by Charles Tanford) refers to the idea that energetically protein folding is driven by two factors:- Hydrophobic side-chains prefer to "get away" from water, whilst. Hydrophilic side-chains prefer to interact with the water. When this is extended to the interaction of ligands with the binding site the classic concept of the hydrophobic effect is as follows: A hydrophobic ligand (or surface on the binding site) disrupts the structure of bulk water and decreases entropy because of stronger bonding and ordering of water molecules around the solute, if ligand and binding site associate then some of the water molecules can be returned to bulk water.
But remember many of the off-target liabilities HERG, CYP interactions, Transporters, have strong correlations with lipophilicity.
Back to Drug Discovery Resources
Updated 7 June 2010