
What is the issue?
- In recent years, a number of drugs have been withdrawn from the market because of reports of sudden cardiac death.
- In all cases, long QT syndrome an abnormality of cardiac muscle repolarization that is characterized by the prolongation of the QT interval in the electrocardiogram, was implicated as inducing ventricular tachycardia that can spontaneously degenerate to ventricular fibrillation and cause sudden death.
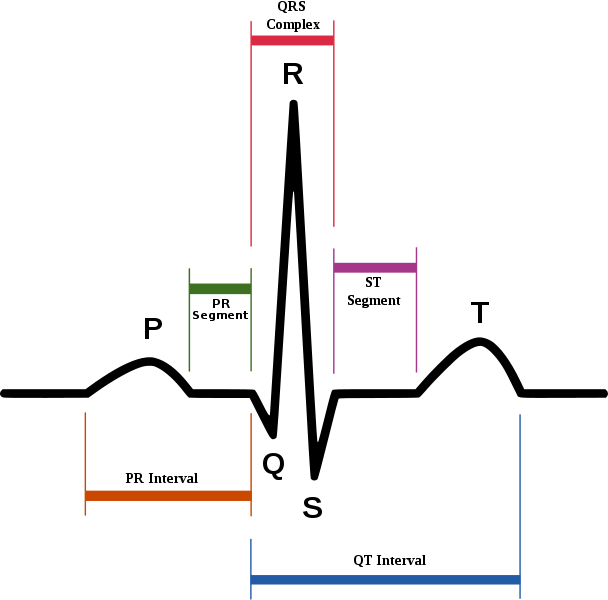
- Virtually every case of a prolonged duration of cardiac action potential related to drug exposure can be traced to one specific mechanism: blockade of IKr current in the heart.
- Blockade of Kv11.1 potassium ion channel encoded by the hERG gene (KCNH2) is widely regarded as the predominant cause of drug-induced QT prolongation. This channel is critical for the repolarisation of the cardiac membrane. hERG forms the major portion of one of the ion channel proteins (the 'rapid' delayed rectifier current (IKr)) that conducts potassium (K+) ions out of the muscle cells of the heart, and this current is critical in correctly timing the return to the resting state of the cell membrane during the cardiac action potential.
- If drug levels of even a modest affinity hERG ligand rise, due to inhibition of a co-administered CYP inhibitor, QT prolongation may be observed
- Since 2005, the FDA has required that new molecular entities are evaluated in a Thorough QT (TQT) study to determine a drug's effect on the QT interval. The TQT study serves to assess the potential arrhythmia liability of a drug. As the pharmaceutical industry has gained experience in performing TQT studies, it has become evident that traditional QT correction formulas such as QTcF, QTcB, and QTcI may not always be suitable for evaluation of drugs impacting autonomic tone.
Typical Inhibitors
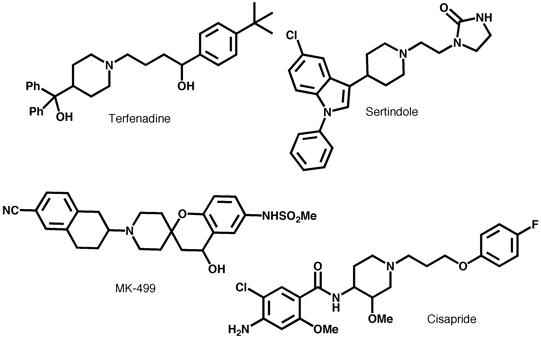
Pharmacophore model consists of a basic nitrogen center flanked by aromatic or hydrophobic groups attached with flexible linkers, receptor modeling and mutagenesis studies highlight interactions between ligand (MK-499) and V625, G648, Y652 and F656. PNAS 97, 12329 (2000) are consistent with the pharmacophore model.
Empirical observations
- Zwitterions very rarely cause problems
- Almost all models include cLogP, a reduction of 1 logP unit in logP often leads to a 0.8 log unit in hERG activity
- Modify pi-Stacking interactions to reduce affinity
- Attenuation of pKa of basic amines, or steric blocking
- Do both, beta-alkoxy/hydroxy reduce pKa and LogP
- Acids less of an issue except for lipophilic acids.
Updated:- Given the issues that many drug discovery programs have had with off-target activity at the hERG channel I've started to compile a database of literature binding data. To date I have information on over 1600 compounds, my aim is to use the data to develop predictive models that I will make freely available, together with the complete data-set which I hope can be used to test novel models.
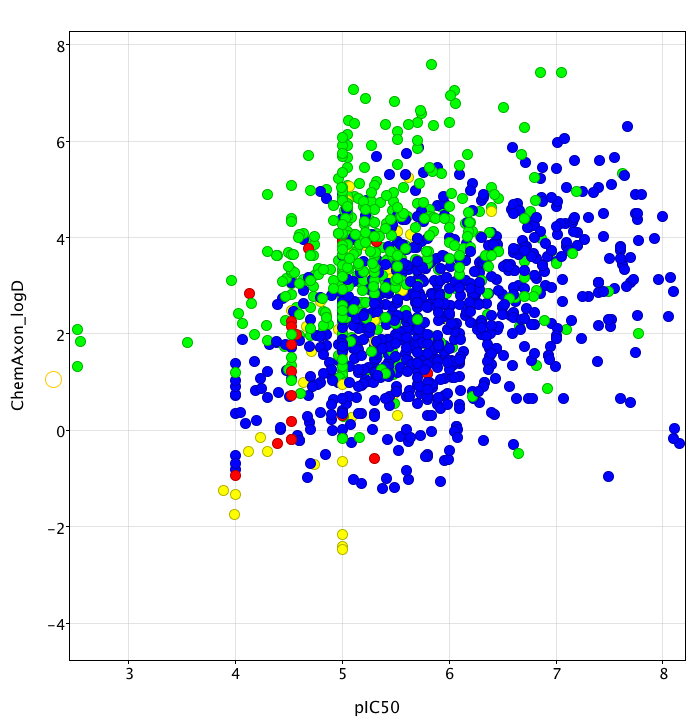
The plot below (created using Vortex)shows pIC50 calculated from the literature IC50 data versus calculated logD determined using the ChemAxon tools the colour coding shows the overall general tend of increasing hERG activity as logP increases with bases (blue) being more prone to HERG activity than neutral (green) for a given lipophilicity. It also highlights the reduced liability seen with acids (red) and zwitterions (yellow).
Whilst the majority of data is derived from radioligand binding experiments (using either Dofetilide or MK-499), there is a substantial amount of data from patch clamp experiments, particularly using the ionworks system. I collated enough data now (covering 6 orders of magnitude) to give an idea of how the assays compare. As you can see there is a reasonable correlation between the assays, but there are one or two outliers. Which is more predictive of in vivo activity is an excellent question that I don’t have the data to answer yet.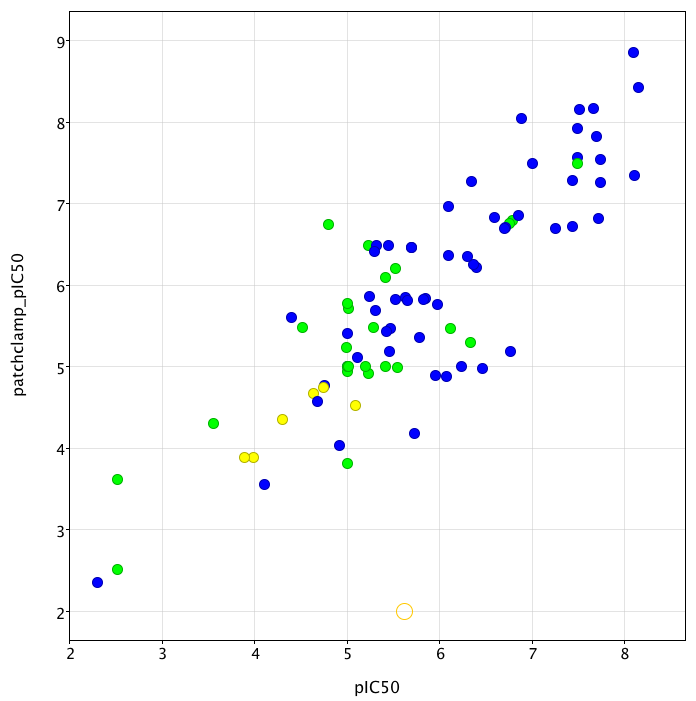
I still need to increase the size of the data-set, and if anyone can direct me to any publicly available data, or to publications that contain data I'd much appreciate it.
What level of selectivity is needed?
In vitro binding affinity does not always correlate with observed in vivo activity
Need to check early in programme
Better to focus in vivo, on a window (10-100 fold) over the observed plasma Cmax at the likely clinical dose, but remember QT prolongation can be seen with low levels of HERG channel occupancy.
S. Kongsamut et al. European Journal of Pharmacology 450 (2002) 37-41 have examined the influence of HERG affinity on QT-prolongation, and found for a series of antipsychotic drugs, the ratio of total plasma concentration to HERG IC50 ([Total plasma]/HERG IC50 )appeared to correspond well with the observed changes in QTc. This ratio ranged from approximately 11 for thioridazine (which displayed an almost 30 ms prolongation in QTc) down to 0.03 for olanzapine (1 ms prolongation in QTc).
Comparison of QTc changes, HERG IC50 and plasma concentrations for various antipsychotic drugs
| Drug | DQTc (ms) | HERG IC50 (nM) | [Total plasma](nM) | [Total plasma]/HERG IC50 |
|---|---|---|---|---|
| Thioridazine | ||||
| Ziprasidone | ||||
| Quetiapine | ||||
| Risperidone | ||||
| Olanzapine |
Worth reading:-
The FDA Guidance for Industry, E14 Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non-Antiarrhythmic Drugs
Medicinal Chemistry of hERG Optimizations: Highlights and Hang-Ups, Jamieson, Journal of Medicinal Chemistry, 2006, 5029
Last Update 9 Feb 2010
Back to Pre-Clinical Toxicity
Back to Drug Discovery Resources