
Nearly all Drug-Drug Interactions (DDI) are due to Phase I enzymes, usually cytochrome P450 enzymes. Many drug interactions are due to the impact one drug can have on the metabolism of a second drug’s metabolism. One major system involved in metabolic drug interactions is the enzyme system comprising the cytochrome P450 oxidases. This system may be affected by either enzyme induction or enzyme inhibition or in some cases both potentially causing significant changes in the plasma levels of administered drugs.
- Enzyme induction - Drug A induces the body to produce more of an enzyme which metabolises drug B. This reduces the effective concentration of drug B, which may lead to loss of effectiveness of drug B. Drug A effectiveness may or may not altered.
- Enzyme inhibition - Drug A inhibits the enzyme metabolising drug B, thus an elevation of drug B occurs possibly leading to an overdose.
- Bioavailability - Drug A influences the absorption of drug B perhaps by blocking active uptake mechanism or inhibiting active transport mediated excretion in the gut.
Cytochrome P450 Inhibition (In Progress)
As might be expected based on the substrate specificities there are wide range of known inhibitors and the structure-activity has been elucidated to some extent.
The X-ray crystal structures are available for some of the isoforms.
CYP 1A2 Potent inhibitors Fluoxamine, Ciprofloxin.
PDB Codes
2HI4 Crystal Structure of Human Microsomal P450 1A2 in complex with alpha-naphthoflavone
CYP 2C9 Potent inhibitors Fluconazole.
PDB codes
1R9O Crystal Structure of P4502C9 with Flurbiprofen bound
1OG5 STRUCTURE OF HUMAN CYTOCHROME P450 CYP2C9
CYP 2C19 Potent inhibitors Omeprazole
CYP 2D6 Potent inhibitors Bupropion, Fluoxetine, Paroxetine, Quinidine.
PDB codes
2F9Q Crystal Structure of Human Cytochrome P450 2D6
CYP 3A4 Potent inhibitors Indinavir, clarithromycin, ketoconazole telithromycin.
PDB codes
2V0M CRYSTAL STRUCTURE OF HUMAN P450 3A4 IN COMPLEX WITH KETOCONAZOLE
2HI4 Crystal Structure of Human Microsomal P450 1A2 in complex with alpha-naphthoflavone
2J0D CRYSTAL STRUCTURE OF HUMAN P450 3A4 IN COMPLEX WITH ERYTHROMYCIN
Cytochrome P450 Inhibitors Pharmacophores
The CYP2D6 inhibitor pharmacophore has been described by Strobel et al, they used the high affinity inhibitor Ajmalicine (3nM) as the template to develop the model.


Based on molecular modeling and enzyme inhibition studies, the pharmacophore was defined by a positively charged nitrogen atom (possibly interacting with ASP301 and/or Glu216)(blue) and a flat hydrophobic region (plane) that maximally extends up to a distance of 7.5 A from this charged nitrogen atom. Compounds showing only these minimum requirements were only weak inhibitors, however. A higher inhibitory potency was observed when additional groups with a negative molecular electrostatic potential and hydrogen bond acceptor properties (red) were present at distances of 4.8-5.5 A and 6.6-7.5 A from the nitrogen atom, respectively. Whilst useful for modeling studies and offering structural insights the data set used is relatively small and probably not sufficient for general screening. More recently Ekins et al have proposed rapid computational filters based on a data set of 1750 molecules using a Random Forest classification. This model correctly identified the most potent inhibitors, however such models tend to be difficult for the bench chemist to interpret the key structural features. There is a recent review of 2D6 drug interactions which emphasizes the importance of the basic center in the ligand (In a data set of 3500 compounds of the 2D6 IC50 < 1uM 92% were basic), lipophilicity (clog P) also resulted in greater CYP2D6 inhibition for basic and neutral compounds (45% of bases with 1 < clog P < 3 exhibited 2D6 IC50 < 10 uM, in contrast 72% of bases with 3 < clog P < 5 exhibited 2D6 IC50 < 10 uM).
Strategies to reduce 2D6 inhibition
For molecules that contain a heteroatom that might coordinate to the iron (imidazoles, pyridines, triazoles, thiadiazoles etc.) substitution adjacent to the nitrogen or modifying the pKa can help. As can be seen in the example below substitution on the thiadiazole thought to bind to the iron has a dramatic effect on inhibition.
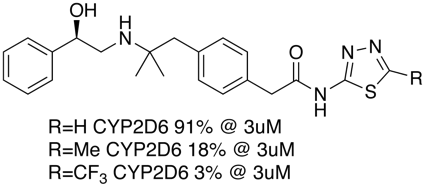
For compounds that don’t bind to the haem iron, reduction of lipophilicity (cLogP) can have a significant effect as shown in the example below.

CYP3A4 is probably the most important enzyme in drug metabolism (J Pharm and Exp Ther 290 (1), 429, 1999). The large lipophilic binding site can accommodate a wide variety of inhibitors. Many diverse known inhibitors, often include heterocycle that binds to the haem iron in the active site, for example the anti-fungal ketoconazole shown below.

A crystal structure of Ketoconazole bound to human CYP3A4 is available, this is the crystal structure 2V0M displayed in moe, ketoconazole is shown in yellow and the haem in red. The nitrogen of the imidazole is bound to the iron, the surface of the protein is coloured coded green= lipophilic, purple= polar. The majority of the binding site is open and lipophilic and even a relatively large molecule like ketoconazole is comfortably accommodated. Other than the coordination to the iron the other major interactions are arene-arene stacking.
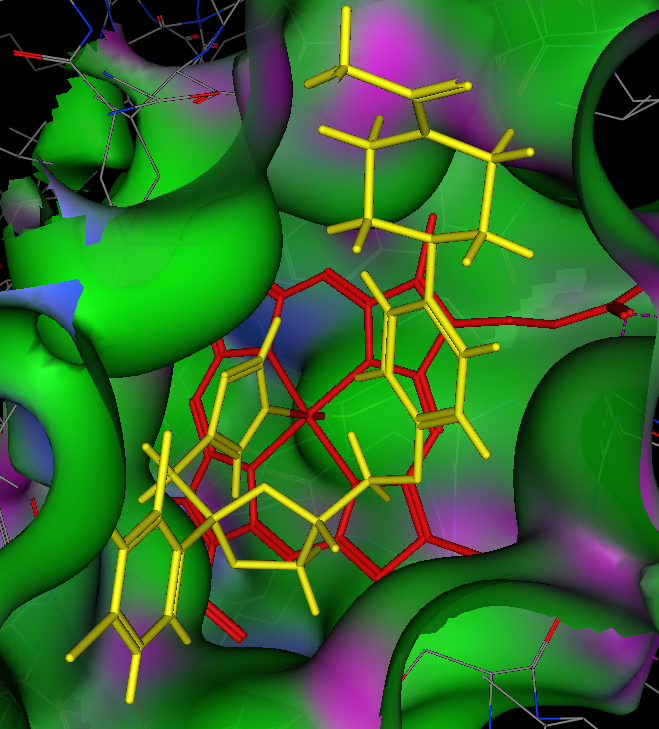
Alternatively click here to view in JMOL 2V0M. You should be able the iron (orange atom) bound to the imidazole nitrogen of ketoconazole.
Strategies to reduce 3A4 inhibition
For molecules that contain a heteroatom that might coordinate to the iron (imidazoles, pyridines, triazoles) substitution adjacent to the nitrogen or modifying the pKa can help. Attempts to reduce inhibition by substitution elsewhere are often futile, the binding site is so large that the molecule simply binds in a slightly different pose. Many computational models have been built and LogD is a term that is frequently highlighted, given the lipophilic nature of the active site it is not surprising that the introduction of polar substitution is often beneficial, this may also reduce the partitioning into the liver. If arene-arene stacking is likely to be important modification of the ring electrostatics can also reduce binding.
Cytochrome P450 Induction
The pregnane X receptor (hPXR) is the major determinant of CYP3A gene regulation by drugs and other xenobiotics. In addition, PXR mediates induction of P450s 2B6, 2C8/9, and 3A7, as well as the drug transporters MDR1, organic anion transporting polypeptide C, bile salt export protein, and multidrug resistance-associated protein 2. Binding site is large and hydrophobic with several important hydrogen bonding interactions. May be multiple binding conformations. Similar to CYP3A pharmacophore, many (but not all) CYP3A substrates/inhibitors are also CYP3A inducers.
Last Update 30 Nov 2009
Back to ADME Properties
Back to Pre-Clinical Toxicity
Back to Drug Discovery Resources