
Fluoxamine COVID-19 treatment
There have been several recent studies suggesting fluvoxamine may be beneficial when treating COVID-19 patients. Most of these reports are small trials that lack the statistical power to provide a solid answer. This has changed with the TOGETHER trial consortium who have been evaluating several different potential treatments shown in the table below. They also undertook a study investigating treatment with Ivermectin that was "stopped for futility".

The results of the Fluvoxamine trial have now been published "Effect of early treatment with fluvoxamine on risk of emergency care and hospitalisation among patients with COVID-19: the TOGETHER randomised, platform clinical trial" DOI. This appears to be a large (741 patients were allocated to fluvoxamine and 756 to placebo) well conducted blinded trial. The average age of the patients was 50 years, 42% male 58% female. Primary end points were related to hospitalisation. They enrolled only participants with diagnosed COVID-19 and less than 7 days of symptom onset using a commercially available COVID-19 rapid antigen test.
The proportion of patients observed in a COVID-19 emergency setting for more than 6 h or transferred to a teritary hospital due to COVID-19 was lower for the fluvoxamine group compared with placebo (79 [11%] of 741 vs 119 [16%] of 756).
The mechanism of action is uncertain but the initial hypothesis was based on the anti-inflammatory action of fluvoxamine. Fluvoxamine is a Selective serotonin reuptake inhibitor (SSRI), however if does have other activities CHEMBL1409/

SMILES = COCCCC/C(=N\OCCN)c1ccc(C(F)(F)F)cc1.O=C(O)/C=C\C(=O)O
It is worth noting Fluvoxamine is not an antiviral but it may act via anti-inflammatory effects that likely stem from its regulation of S1R, which modulates innate and adaptive immune responses and Fluvoxamine may serve to dampen cytokine storms. There have been reviews of the potential mechanism of action DOI.
Antiviral Molnupiravir Reduced the Risk of Hospitalization or Death by Approximately 50 Percent Compared to Placebo for Patients with Mild or Moderate COVID-19
Merck have just announced that the Investigational Oral Antiviral Molnupiravir Reduced the Risk of Hospitalization or Death by Approximately 50 Percent Compared to Placebo for Patients with Mild or Moderate COVID-19 in Positive Interim Analysis of Phase 3 Study.
Great news, but bear in mind this is only an interim study, so keep those fingers crossed.
Molnupiravir (development codes MK-4482 and EIDD-2801) is an experimental antiviral drug which is orally active and was developed for the treatment of influenza. It is a prodrug of the synthetic nucleoside derivative N4-hydroxycytidine, and exerts its antiviral action through introduction of copying errors during viral RNA replication.

InChI=1S/C13H19N3O7/c1-6(2)12(19)22-5-7-9(17)10(18)11(23-7)16-4-3-8(15-21)14-13(16)20/h3-4,6-7,9-11,17-18,21H,5H2,1-2H3,(H,14,15,20)/t7-,9-,10-,11-/m1/s1
CHEBI https://www.ebi.ac.uk/chebi/searchId.do?chebiId=CHEBI:180653
COVID moonshot to get £8M funding
A great start to the week.
The COVID Moonshot, a non-profit, open-science consortium of scientists from around the world dedicated to the discovery of globally affordable and easily-manufactured antiviral drugs against COVID-19 and future viral pandemics has received key funding of £8 million from Wellcome, on behalf of the Covid-19 Therapeutics Accelerator.
Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2
An interesting open access paper in Science "Drug-induced phospholipidosis confounds drug repurposing for SARS-CoV-2" DOI points a potential flaw in interpreting in vitro data.
Repurposing drugs as treatments for COVID-19 has drawn much attention. Beginning with sigma receptor ligands, and expanding to other drugs from screening in the field, we became concerned that phospholipidosis was a shared mechanism underlying the antiviral activity of many repurposed drugs. For all of the 23 cationic amphiphilic drugs tested, including hydroxychloroquine, azithromycin, amiodarone, and four others already in clinical trials, phospholipidosis was monotonically correlated with antiviral efficacy. Conversely, drugs active against the same targets that did not induce phospholipidosis were not antiviral.
Phospholipidosis is well known phenomena for those involved in drug discovery, it is based on the physicochemical properties of the molecule and results in excessive accumulation of intracellular phospholipids in tissues, such as the liver, kidney and lung. The resulting accumulation can lead to liver, kidney, or respiratory failure.
Drug-induced phospholipidosis can be determined by measuring the accumulation of specific fluorescent probes in HepG2 cells or primary hepatocytes.
There is a more detailed explanation of the consequences here, "A Strategy for Risk Management of Drug-Induced Phospholipidosis" DOI.
LifeArc funding Project Moonshot
The independent medical charity LifeArc is to support an international effort to rapidly develop a potential antiviral candidate for COVID-19.
The grant will be entirely dedicated to the COVID Moonshot initiative, which PostEra jointly leads alongside leading scientists from Diamond Light Source, Oxford University, Weizmann Institute, Memorial Sloan Kettering Cancer Center and MedChemica.
21st RSC / SCI Medicinal Chemistry Symposium
Registration for the 21st RSC / SCI Medicinal Chemistry Symposium Monday-Wednesday, 13th-15th September 2021 hosted at Churchill College, Cambridge, UK is now open. Twitter hashtag - #CamMedChem21
As usual there is a stella lineup of presentations and there is still time for some submissions.
I'd like to highlight one talk that is close to my heart. Ed Griffen will talking about The COVID Moonshot: SARS-CoV2 oral antiviral therapeutics from an Open Science global collaboration.
The COVID Moonshot is an ambitious crowdsourced initiative to accelerate the development of a COVID antiviral. We work in the open with no intellectual property constraints. This way, any scientist can view submitted drug designs and experimental data to inspire new design ideas. We use our cutting-edge machine learning tools and Folding@home's crowdsourced supercomputer to determine which drug designs to send to our partners to make and test in the lab. With each drug design tested, we get closer to our goal.
This talk will be in the late breaker session and because of the open nature of the project it will be a chance to really see the very latest results "hot off the press".
You can have a look at the latest results here now. https://covid.postera.ai/covid.
30th April: Late breaker and early poster deadline
23rd July: Final poster deadline
Registration link https://www.maggichurchouseevents.co.uk/bmcs/cmc21/index.htm
Cross-referencing the Project Moonshot compounds
The project COVID moonshot is generating a significant amount of data both biochemical data distributed by PostEra and crystallographic data generated and distributed by the team at Diamond.
The COVID Moonshot is an ambitious crowdsourced initiative to accelerate the development of a COVID antiviral. We work in the open with no intellectual property constraints. This way, any scientist can view submitted drug designs and experimental data to inspire new design ideas. We use our cutting-edge machine learning tools and Folding@home's crowdsourced supercomputer to determine which drug designs to send to our partners to make and test in the lab. With each drug design tested, we get closer to our goal.
It is sometimes difficult to cross-reference compounds between multiple sources so I've downloaded the compounds with associated data calculated InChiKeys and then used the InChiKey to link compounds from different sources within Vortex. This means you have the biochemical data together with PDB code (if available) or the fragalysis code for the crystal structure. I've also annotated with identifiers from multiple databases (ChEMBL, PubChem etc.), calculated physicochemical properties (LogP/D, TPSA, HBD/A etc) and then exported in sdf format. I've also clustered the structures to aid navigation.
You can download the zipped sdf file here.
Updated. I was asked if I could provide this file in SMILES format so here it is.
I plan to try and have a look at ways to visualise the data when I can find some free time.

The SARS-CoV-2 main protease as drug target
A very useful primer for those interested in contributing to the ongoing research efforts at COVID moonshot and Open Source COVID-19.
The SARS-CoV-2 main protease as drug target DOI
The unprecedented pandemic of the severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is threatening global health. The virus emerged in late 2019 and can cause a severe disease associated with significant mortality. Several vaccine development and drug discovery campaigns are underway. The SARS-CoV-2 main protease is considered a promising drug target, as it is dissimilar to human proteases. Sequence and structure of the main protease are closely related to those from other betacoronaviruses, facilitating drug discovery attempts based on previous lead compounds. Covalently binding peptidomimetics and small molecules are investigated. Various compounds show antiviral activity in infected human cells.
Remember a hit in a screen is just the very first step, there is much more to consider before it can be described as a drug candidate.
Another PYMOL session file
A full-length model of glycosylated SARS-Cov-2 spike protein is recently described in literature by Casalino et. al. The PDB files for models are available at https://amarolab.ucsd.edu/covid19.php. These PDB files contain data for spike protein, glycans, lipid membrane, ions, and solvent.
Manish Sud has generated an annotated PyMol session file to view the model of spike protein present in open state conformation. The PyMOL session file is quite helpful during the reading of the article describing the work. It's a bit of elbow grease work to set up appropriate views in PyMOL and Manish has kindly shared it. It's available for download at http://www.mayachemtools.org/Download.html. I'm sure many will find it helpful.

The size of uncompressed PyMOL session file is quite large. It might take few minutes to load it into PyMOL, based on your hardware specifications.
Manish has also provided session files for SARS-CoV-2 Mpro ligands.
More COVID-19 MPro Activity Data
One of the best drug targets among coronaviruses is the main protease (Mpro), this enzyme is essential for processing the polyproteins that are translated from the viral RNA and the recognition sequence at most sites is Leu-Gln↓(Ser,Ala,Gly) and since no human enzymes have similar specificity inhibitors should be very specific. Mpro is a papain-like protease cysteine protease.
I've previously described the fragment hits from a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Full details of the screening effort are described here https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Downloads.html
Additional biological results from project moonshot are now available. You can browse the data here https://postera.ai/covid/activity_data.
These results contain a significant milestone with the identification of the first sub micromolar non-covalent inhibitor.
JAG-UCB-a3ef7265-20 has been titrated twice now and has an IC50 of 0.6 uM.

This compound is a racemic mixture and the synthesis of the individual enantiomers is underway, if the activity predominantly lies with a single enantiomer we could see a further improvement in activity. The original submission was based on a pharmacophore search of Enamine based on amino-pyridine hits. I highlight this to underline the importance of simple descriptor-based searches, they are often highly competitive with sophisticated docking studies and require orders of magnitude less compute resources.
Since this research is being conducted in the public domain a number of other people have been able to contribute further ideas based on this exciting discovery.

Dexamethasone shown to reduce COVID-19 mortality
The NIHR-funded and supported study RECOVERY (Randomised Evaluation of COVid-19 thERapY) has announced that the steroid dexamethasone has been identified as the first drug to improve survival rates in certain coronavirus patients.
A total of 2104 patients were randomised to dexamethasone once per day for ten days and were compared with 4321 patients randomised to usual care alone. Among the usual care control group, 28-day mortality was highest in those on ventilators (41%), intermediate in those on oxygen only (25%), and lowest among those who were not receiving any respiratory intervention (13%).
The study, conducted at the University of Oxford and led by Professor Peter Horby and Professor Martin Landray, found that dexamethasone reduced the risk of dying by one-third in ventilated patients and by one fifth in other patients receiving oxygen only. There was no benefit among those who did not need respiratory intervention.
Dexamethasone is an inexpensive corticosteroid medication used to treat many inflammatory and autoimmune conditions, such as rheumatoid arthritis and bronchospasm. Glucocorticoids are part of the feedback mechanism in the immune system, which modulates certain aspects of immune function. They bind to the glucocorticoid receptor, and the activated complex up-regulates the expression of anti-inflammatory proteins and represses the expression of proinflammatory proteins.

Dexamethasone CHEMBL384467 has good oral bioavailability (80-90%) and a reasonable half-life (4 h), with a high volume of distribution (> 50L). It is also available as a 3.3 mg/mL solution for intravenous use. Dexamethasone is extensively metabolised to 6-hydroxydexamethasone via CYP3A4 mediated oxidation.
The oral LD50 in female mice is reported to be 6.5g/kg.
Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease
Full details of the Crystallographic and electrophilic fragment screening of the SARS-CoV-2 main protease are now published. https://www.biorxiv.org/content/10.1101/2020.05.27.118117v1.full.pdf
An extraordinary effort highlighted by the timeline shown below.

The results of the first round of biological results from project moonshot are in. You can browse the data here https://postera.ai/covid/activity_data.
A listing of my posts on COVID-19 are here.
First round of MPro bioactivity results
One of the best drug targets among coronaviruses is the main protease (Mpro), this enzyme is essential for processing the polyproteins that are translated from the viral RNA and the recognition sequence at most sites is Leu-Gln↓(Ser,Ala,Gly) and since no human enzymes have similar specificity inhibitors should be very specific. Mpro is a papain-like protease cysteine protease
I've previously described the fragment hits from a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Full details of the screening effort are described here https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Downloads.html
The results of the first round of biological results from project moonshot are in. You can browse the data here https://postera.ai/covid/activity_data.

Most of the most active compounds are chloroketones or acrylamides, presumably covalent inhibitors, and they all show selectivity over Trypsin (IC50 >99 uM).
There are a few structures that look more like competitive inhibitors shown below

A number of these structures now have crystal structures available.
A sdf file containing these non-covalent structures is here
Fantastic work by all involved.
COVID-19 and the Identification of "Drug Candidates"
One of the really heartening things to come out of the current pandemic is the willingness of many scientists to put aside their own research and throw themselves into the efforts to find a treatment. However, lack of domain expertise is always a problem when scientists enter a new field, so I thought I'd put together a few things to consider.
In silico screening, for docking experiments you need to put considerable effort into ensuring the protein structure used is appropriate, you can't simply download a PDB file from the Protein Data Bank and use it. It will undoubtedly contain errors, you will need check protonation, hydrogen bonds etc. Then there is the issue of deciding which solvent molecules are important. Binding energies, docking scores are not as accurate as many seem to assume and no substitute for an experienced medicinal chemists looking at the bound poses, I've tried to summarise the types of molecular interactions here. Remember to also think about the impact of solvation. For other virtual screening approaches you need to be very careful about the quality of the input data. In many cases it will be heavily biased towards actives.
In silico predictions are no substitute for biological data, if you are using repurposed drugs or available chemicals there is really no excuse for not generating the appropriate in vitro biological data, there are many labs who would be happy to collaborate. If the molecules are novel many custom synthesis companies have offered to help. Remember that the IC50 is probably not that useful, it is likely that you will want to block the target 100% so you need to be above the IC95. In vitro biochemical assays using isolated enzymes will often give a false sense of potency, you should also determine activity in a cell-based assay in the presence of plasma.
If you are proposing a repurposed drug there will be a lot of information about the drug in the public domain, you may well need to search for compound codes, and various drug name synonyms. UniChem is a very useful web service for cross-referencing between chemical structure identifiers.
There are now many free, web-accessible databases some useful starting points are shown in the table below.
| Name | Link | Description |
|---|---|---|
| ChEMBL | https://www.ebi.ac.uk/chembl/ | A database of bioactive drug-like small molecules, it contains 2-D structures, calculated properties (e.g. logP, Molecular Weight, Lipinski Parameters, etc.) and abstracted bioactivities (e.g. binding constants, pharmacology and ADMET data). |
| PubChem | https://pubchem.ncbi.nlm.nih.gov | Three linked databases within the NCBI's Entrez information retrieval system. These are PubChem Substance, PubChem Compound, and PubChem BioAssay. Many compounds have links to primary literature and patents |
| Guide to Pharmacology | https://www.guidetopharmacology.org/GRAC/searchPage.jsp | An expert-driven guide to pharmacological targets and the substances that act on them. |
| DrugBank | https://www.drugbank.ca | The DrugBank database is a unique bioinformatics and cheminformatics resource that combines detailed drug data with comprehensive drug target information |
| NCI Thesaurus | https://ncithesaurus.nci.nih.gov/ncitbrowser/ | NCI Thesaurus (NCIt) provides reference terminology for many NCI and other systems. It covers vocabulary for clinical care, translational and basic research, and public information and administrative activities |
| Clinical Trials | https://clinicaltrials.gov | A database of privately and publicly funded clinical studies conducted around the world |
| FDA | https://www.fda.gov | Food and Drug Administration responsible for safety and efficacy of drugs |
| WIPO | https://www.wipo.int/portal/en/index.html | World IP services |
Find out the original target and mode of action. I've seen a couple of proposed compounds that are known prodrugs, the parent compound is designed to either breakdown or be modified in vivo to yield the active compound. The prodrug may have negligible systemic exposure. Covalent modifiers may look attractive but selectivity is always a concern and they may have narrow therapeutic windows.
Look at the original indication, many anticancer drugs are extremely toxic and could not be given other patients. Similarly, drugs that reduce blood pressure or other physiological changes may be problematic. You may well be able to find counter-screening data, this could highlight problematic off-target activities.
Look at the approved dosing regime, if a drug is only approved for doses of 2 ug/kg there might well be good reasons, and if your proposed drug only has uM activity in the in vitro assays you won't be able to generate sufficient plasma concentrations. Check what safety studies have been undertaken, are they sufficient to support multi-day dosing?
Look at the pharmacokinetics, you should be able to model the dosing regime needed to maintain plasma concentrations above IC95, this will may need to be maintained 24 hours a day. Check protein binding and distribution and use in the predictive modelling.

Look for the routes of administration, for in intensive care I suspect many will need the drug to be administered i.v. if there is no intravenous formulation is the drug soluble enough for one to be developed, ber in mind the limitations of intravenous formulations
Many of the patients will be on multiple drugs, both to treat the viral infection but also adventitious bacterial infections and since many are elderly and have pre-existing medical conditions they may have a cocktail of drugs prescribed. Drug-Drug interactions thus become a major concern, any proposed drug to treat the virus that has major interactions with CYP450 enzymes (induction, inhibition or metabolism) is likely to hugely complicate the overall dosing regime.
Check for any toxicity information, particularly black box warnings. HERG inhibition and QT prolongation is an issue that most drug discovery projects have to address at some point. This is particularly worrying if coupled with potential drug-drug interaction described above. You should also be able to find the data from safety studies, these may describe the dose limiting toxicities.
All of this information should be in the public domain, and if you are proposing a compound as a "Drug Candidate" you should not be expecting someone else to pull it all together to decide whether it is worth pursuing clinically.
Updated 26 April 2020
This Week in Virology
An interesting weekly podcast that is currently topical.
This week Doris Cully joins TWiV to discuss inhibition of SARS-CoV-2 in cell culture by ivermectin.
COVID-19 Registered Trials
There are now a number of clinical trials underway and this review by The Centre for Evidence-Based Medicine provides an excellent summary of the trials that are taking place. They describe proposed pharmacological interventions and their mechanisms, when known, but unfortunately don't give the chemical structures.
Updated
I've also now included a few other structures that people have sent to me.
Here is the workflow I use to get the structures and access more information about the compounds.
Create a text file with all the structures mentioned
ASC09
Azvudine
Azithromycin
Baloxavir
Carriomycin
Chloroquine
cobicistat
Danoprevir
Darunavir
Dihydroartemisinin
Favipiravir
Fingolimod
hydroxychloroquine
Jakotinib
Leflunomide
Lopinavir
marboxil
Methylprednisolone
oseltamivir
piperaquine
Remdesivir
ribavirin
ritonavir
Ruxolitinib
Suramin
Thalidomide
thymosin
Triazavirin
Umifenovir
Now read the text file into Vortex

The use a Name to Structure script to use a web service to get the structures, in this case I used ChemSpider. Now generate the InChiKey from the structures.

We can now use the InChiKey to search UniChem using another Vortex script to get identifiers for the molecule from various databases.
UniChem efficiently produces cross-references between chemical structure identifiers from different databases

We can then use the identifiers to search the various databases for more information
I've been asked if I could provide the structures for download
Here it is in SDF file format http://cambridgemedchemconsulting.com/news/files/COVID19/coviddata.sdf.zip
And in SMILES format http://cambridgemedchemconsulting.com/news/files/COVID19/forpost.smi.zip.
The quality of the crystal structure is critical
Crystal structures are not perfect, and it is important to understand the limitations and not assume as Derek Lowe once put it, they are a "message from God". It might be worth reading the section on structure-based design.
With this in mind I thought I'd flag this message from Bobby Glen (Cambridge) here.
Hi, we’re still (Jason at CCDC) porting GOLD to our HPC system so we can basically parallel dock. We should be able to dock and score early next week I hope, There are a few issues we also are addressing wrt the crystal structures, Gerard Bricogne at Global Phasing is kindly re-refining the published structure from the ED, this hopefully will inform us of for making some changes to the orientation/pKa and tautomers of the histidines and some of the other AAs. It’s very difficult to ‘see’ hydrogen in x-ray and these are inferred from the structure. We need to be sure we have a decent model of this (at physiological pH) before doing all the calculations. An example is H163, which is in the binding site, and is critical to a few of the interactions seen in ligands for this class of proteases. Automated hydrogen addition can be problematic.
Help design inhibitors of the SARS-CoV-2 main protease
Are you a medicinal chemist currently locked out of your lab?
Why not take a break from writing papers/reports and lend your expertise to this effort, https://covid.postera.ai/covid. They have identified 60 fragment hits and are asking for insight in what should be made next.
We are now asking for your help in designing new inhibitors based on these initial fragment hits: the exceptionally dense readout suggests countless opportunities for growing and merging, and we need many sharp brains to sift through them; it is also what makes us believe that potency can be directly achieved.
The first round of submissions will be reviewed tonight and the selected molecules will be made by Enamine.
Structures of SARS-CoV-2 ligands PYMOL session files
One of the best drug targets among coronaviruses is the main protease (Mpro), this enzyme is essential for processing the polyproteins that are translated from the viral RNA and the recognition sequence at most sites is Leu-Gln↓(Ser,Ala,Gly) and since no human enzymes have similar specificity inhibitors should be very specific. Mpro is a papain-like protease cysteine protease
I've previously described the fragment hits from a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Full details of the screening effort are described here https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Downloads.html
I've downloaded all the structures that were screened, both those that bind and those where no binding was observed and put them into a single file, also added inChiKey, SMILES, PubChem ID, PDB ID of ligand if known and a range of other identifiers from different databases, the file is available here http://cambridgemedchemconsulting.com/news/files/Archive.zip.
Whilst that is probably sufficient for those looking at cheminformatics driven approaches to designing new molecules anyone wanting to undertake structure based design would need to download all the structures and then overlay them to visualise on their desktop. Fortunately Manish Sud of MayaChemTools has done the hard work and generated a series of PYMOL session files that allow you explore the enzyme crystal structure and the screening data interactively.
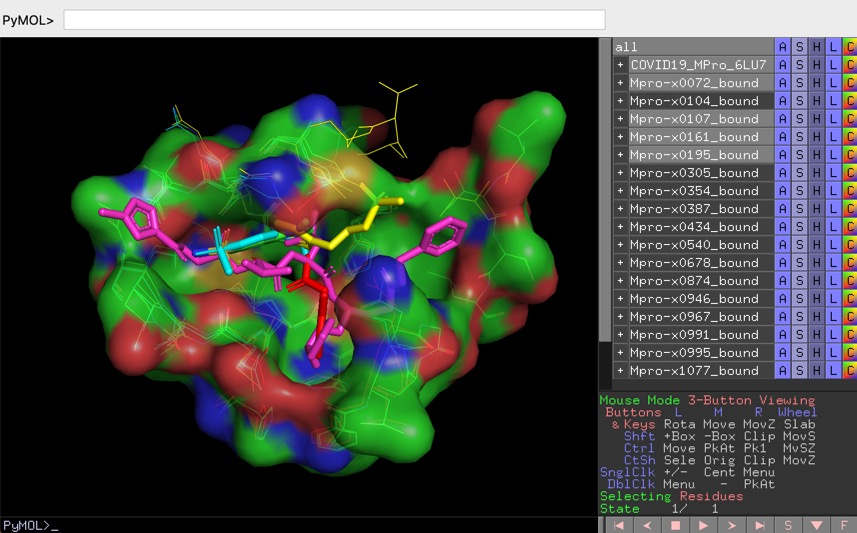
PYMOL is an open source molecular visualisation application, you can download it here https://pymol.org/2/ or install using conda
conda install -c schrodinger pymol
If you have not used it before there is a tutorial here
The PyMOL session files are setup to facilitate the analysis of protein ligand interactions in the binding pocket, to view the files select "Open" from the file menu bar, some of the larger files make take a little while to load.
X-ray crystal structures and electron densities
COVID-19 main protease with unliganded active site (2019-nCoV, coronavirus disease 2019, SARS-CoV-2) 6Y84 and the crystal structure of COVID-19 main protease in complex with an inhibitor N3 6LU7.
The PYMOL session files (zipped) can be downloaded here
http://cambridgemedchemconsulting.com/news/files/COVID19/COVID19-MPro-6LU7-6Y84.pse.zip
Structures for non-covalent ligands
The structures of the non-covalent ligands are here.
If you are not familiar with fragment-based screening there is an introduction here including some examples of fragment growing.
It is likely that fragments will only have very modest affinity and that to completely suppress the enzyme it will require very high affinity ligands with good pharmacokinetics to achieve 100% occupancy for 24 hours per day. For this reason molecules that irreversibly bind to the enzyme might be an attractive alternative option.
Structures for covalent ligands are here
The session file containing the covalent ligands are here
This is a large file so Manish has divided it.
Whilst much of drug discovery deals with non-covalent, reversible interactions with the target protein there are also a class of therapeutic agents that bind covalently to the target protein, these are described on this page. To mitigate the risk of off-target toxicity you will need to maximise the selectivity for the target enzyme. Glutathione conjugation can be used as a surrogate for off-target reactivity.
Getting designs made
Once you have designed a novel ligand have a look at Design a Compound, We Will Make It
Designs will be prioritized by factors, such as ease of synthesis, and toxicity modeling, then synthesized by Enamine and tested by groups around the world. PostEra will be running machine learning algorithms in the background to triage suggestions and generate synthesis plans to enable a rapid turnaround. You will be informed of the progress of the molecules through the main stages (validation, synthesis and testing).
COVID-19 Open Research Dataset Challenge (CORD-19)
There are a number of COVID-19 Kaggle challenges open at the moment, https://www.kaggle.com/datasets?search=COVID.
One of the more recent is:-
COVID-19 Open Research Dataset Challenge (CORD-19)
There is a large body of research and literature continuously evolving around COVID-19. Help the research community and global organizations better digest this to answer key questions."
In response to the COVID-19 pandemic, the White House and a coalition of leading research groups have prepared the COVID-19 Open Research Dataset (CORD-19). CORD-19 is a resource of over 29,000 scholarly articles, including over 13,000 with full text, about COVID-19, SARS-CoV-2, and related coronaviruses. This freely available dataset is provided to the global research community to apply recent advances in natural language processing and other AI techniques to generate new insights in support of the ongoing fight against this infectious disease. There is a growing urgency for these approaches because of the rapid acceleration in new coronavirus literature, making it difficult for the medical research community to keep up.
You can read more about it here
favipiravir shows promise in treatment of COVID-19
Favipiravir, also known as T-705, Avigan, or favilavir is a drug designed to treat RNA viral infections DOI and DOI. It is phosphoribosylated by cellular enzymes to its active form, favipiravir-ribofuranosyl-5'-triphosphate (RTP) and inhibits the RNA-dependent RNA polymerase.

Favipiravir has recently been reported to be effective in the treatment of coronavirus patients Link. It appears to be effective in patients showing mild to moderate symptoms, but not effective in patients showing more severe symptoms.
A search of UniChem using the InChiKey gives details of all identifiers and links to clinical studies.
A number of clinical trials have been completed or are ongoing on ClinicalTrials.gov and can be found here.
Whilst it appears to be safe and well tolerated, and it has been approved for flu it has not yet been approved for COVID-19,
Fragment hits for SARS-CoV-2
A group of researchers including Dave Stuart, Martin Walsh, and Frank von Delft (Diamond Light Source) has performed a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Even before fully analyzing all of the data they have released interim results https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem.html.
The hits can be viewed on fraglaysis here.
I've downloaded all the structures that were screened, both those that bind and those where no binding was observed and put them into a single file, also added inChiKey, SMILES, PubChem ID, PDB ID of ligand if known and a range of other identifiers from different databases, the file is available here http://cambridgemedchemconsulting.com/news/files/Archive.zip

Potential 2019-nCoV 3C-like Protease Inhibitors
Chris Southan recently flagged a number of publications describing possible treatments for 2019-nCoV using repurposed existing drugs "Therapeutic options for the 2019 novel coronavirus (2019-nCoV)" DOI. In addition a recent preprint "Potential 2019-nCoV 3C-like Protease Inhibitors Designed Using Generative Deep Learning Approaches" DOI highlighted the design of potential protease inhibitors. The authors provide the structures of the molecules in the supplementary informations.
I downloaded the sdf file and used a Jupyter notebook to calculate a range of physicochemical properties, the results are shown in the plot below.

As often seen with protease inhibitors, the molecular weight is rather high with the majority of compounds having Mol Weight >500. The calculated LogP is mainly in the range 2 to 5, however because 40% of the molecules are predicted to be basic the LogD is mainly in the range 0-4. This combination of high molecular weight and rather high LogP is likely to compromise the developability score (for more details on develop ability score read "20 years Rule of Five" report here.

The molecules are also predicted to have a rather high number of hydrogen bond donors and acceptors, this contributes to high polar surface area (TPSA). In general TPSA >120 are often associated with poor oral bioavailability, however it should be noted that the TPSA was not calculated on a 3D structure and it is possible that intramolecular hydrogen bonds may reduce the actual TPSA, also described in the 20 years Rule of Five report.
Scanning through the molecules I noticed a number of functional groups that might be a concern (e.g. Micheal Acceptors), I ran a couple of Vortex scripts that flag potential problematic groups based on SMARTS taken from the following publications
http://pubs.acs.org/doi/abs/10.1021/jm901137j
http://pubs.acs.org/doi/abs/10.1021/jm5019093
https://doi.org/10.1177/1087057116639992
https://dx.doi.org/10.1177%2F1087057113516861
I also ran a couple scripts that flag potential liver toxicity or HERG liabilities. These flags should not be used to exclude molecules but should be used to flag molecules for checking experimentally. The script identifies the functional group that has been flagged as a liver toxicity liability, and identifies the most similar molecule in ChEMBL that has HERG activity. The results are shown in the image below.

I also added InChiKeys for better cross referencing.
I've exported all results as an sdf file which can be found here http://cambridgemedchemconsulting.com/news/files/INSCoV_2020sdfv1addedflags.sdf.zip.