
Structures of SARS-CoV-2 ligands PYMOL session files
One of the best drug targets among coronaviruses is the main protease (Mpro), this enzyme is essential for processing the polyproteins that are translated from the viral RNA and the recognition sequence at most sites is Leu-Gln↓(Ser,Ala,Gly) and since no human enzymes have similar specificity inhibitors should be very specific. Mpro is a papain-like protease cysteine protease
I've previously described the fragment hits from a fragment screen against crystals of the main protease (MPro) of SARS-CoV-2, the virus that causes COVID-19. Full details of the screening effort are described here https://www.diamond.ac.uk/covid-19/for-scientists/Main-protease-structure-and-XChem/Downloads.html
I've downloaded all the structures that were screened, both those that bind and those where no binding was observed and put them into a single file, also added inChiKey, SMILES, PubChem ID, PDB ID of ligand if known and a range of other identifiers from different databases, the file is available here http://cambridgemedchemconsulting.com/news/files/Archive.zip.
Whilst that is probably sufficient for those looking at cheminformatics driven approaches to designing new molecules anyone wanting to undertake structure based design would need to download all the structures and then overlay them to visualise on their desktop. Fortunately Manish Sud of MayaChemTools has done the hard work and generated a series of PYMOL session files that allow you explore the enzyme crystal structure and the screening data interactively.
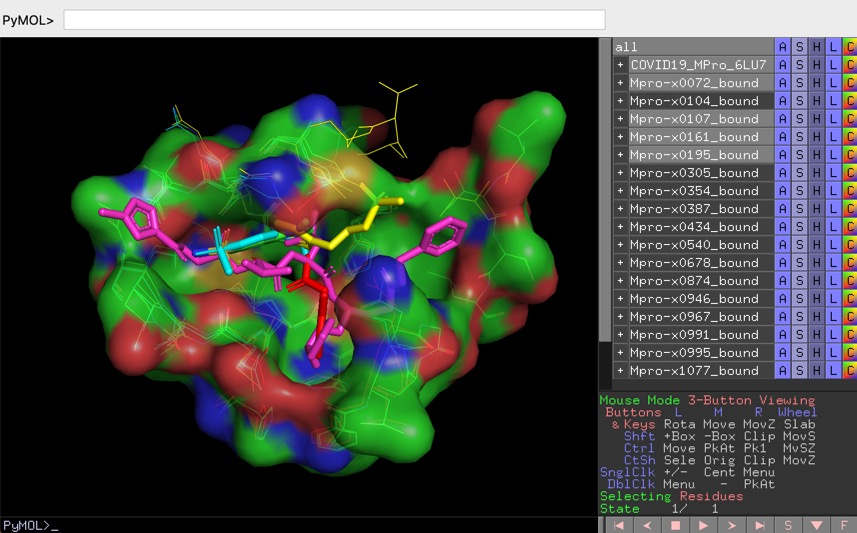
PYMOL is an open source molecular visualisation application, you can download it here https://pymol.org/2/ or install using conda
conda install -c schrodinger pymol
If you have not used it before there is a tutorial here
The PyMOL session files are setup to facilitate the analysis of protein ligand interactions in the binding pocket, to view the files select "Open" from the file menu bar, some of the larger files make take a little while to load.
X-ray crystal structures and electron densities
COVID-19 main protease with unliganded active site (2019-nCoV, coronavirus disease 2019, SARS-CoV-2) 6Y84 and the crystal structure of COVID-19 main protease in complex with an inhibitor N3 6LU7.
The PYMOL session files (zipped) can be downloaded here
http://cambridgemedchemconsulting.com/news/files/COVID19/COVID19-MPro-6LU7-6Y84.pse.zip
Structures for non-covalent ligands
The structures of the non-covalent ligands are here.
If you are not familiar with fragment-based screening there is an introduction here including some examples of fragment growing.
It is likely that fragments will only have very modest affinity and that to completely suppress the enzyme it will require very high affinity ligands with good pharmacokinetics to achieve 100% occupancy for 24 hours per day. For this reason molecules that irreversibly bind to the enzyme might be an attractive alternative option.
Structures for covalent ligands are here
The session file containing the covalent ligands are here
This is a large file so Manish has divided it.
Whilst much of drug discovery deals with non-covalent, reversible interactions with the target protein there are also a class of therapeutic agents that bind covalently to the target protein, these are described on this page. To mitigate the risk of off-target toxicity you will need to maximise the selectivity for the target enzyme. Glutathione conjugation can be used as a surrogate for off-target reactivity.
Getting designs made
Once you have designed a novel ligand have a look at Design a Compound, We Will Make It
Designs will be prioritized by factors, such as ease of synthesis, and toxicity modeling, then synthesized by Enamine and tested by groups around the world. PostEra will be running machine learning algorithms in the background to triage suggestions and generate synthesis plans to enable a rapid turnaround. You will be informed of the progress of the molecules through the main stages (validation, synthesis and testing).