
ENABLE-2 Incubator: Hit validation
ENABLE-2 offers evaluation of the antibiotic potential of novel compounds.
(i) Antibacterial activity (MIC) will be measured against selected Gram-negative and Gram-positive species to identify Hits (defined as compounds with wild-type whole cell activity on at least a species of interest).
(ii) Hits will be evaluated for in vitro cytotoxicity.
(iii) Limited hit expansion to explore SAR is possible if required by the programme and if ENABLE-2 resources are available.
Researchers at publicly funded universities and research institutes in Europe (including non-EU countries such as UK, Norway, Switzerland etc.) are eligible.
ENABLE-2 bacterial species of interest for Hit to Lead development
E.coli, K. pneumoniae, P. aeruginosa, A. baumannii, S. aureus, E. faecium.
UKRI funding to tackle antimicrobial resistance.
Transdisciplinary research to tackle antimicrobial resistance. Total fund £15,000,000.
You must be employed by a research organisation eligible to apply for UK Research and Innovation (UKRI) funding.
Your team and research project will bring new perspectives crossing Councils’ remits to understand and provide solutions to tackle AMR.
The full economic cost (FEC) of your project can be up to £3,000,000. UKRI will fund at 80% of the FEC.
The duration of the award is up to five years.
Covalent Inhibitors
Covalent Inhibitorrs are an increasingly investigated class of drugs. The attraction is the irreversible nature of the binding and subsequent prolonged target engagement. An invaluable resource for this area of research is the Covalent Inhibitor Database that has recently been updated to version 2.0.
This updated version includes 8303 inhibitors and 368 targets, supplemented by 3445 newly added cocrystal structures, providing detailed analyses of non-covalent interactions.
The article describing the database is here DOI.
New approaches to the treatment of Parkinson’s
The website the the RSC-BMCS conference on New approaches to the treatment of Parkinson’s is now live! Details are on the website including registration details.
https://www.rscbmcs.org/events/parkinsons25/
In the dynamic field of drug discovery, the search for new treatments for Parkinson’s disease has never been more critical. It is estimated 10 million people worldwide are living with Parkinson’s – a chronic, progressive neurodegenerative condition resulting from the loss of the dopamine-containing cells of the substantia nigra. Motor and non-motor symptoms of the condition are wide ranging and current treatments only help to manage a small subset of the symptoms and do not modify the disease progression. But there is a wealth of research to find new and better therapeutics for Parkinson’s.
This meeting, specifically designed for professionals dedicated to developing new therapeutics for Parkinson’s disease, will highlight current research offering symptomatic relief and disease modifying approaches.
Abstract submissions for Oral and posters are now open and the submission for is here.
https://www.rscbmcs.org/wp-content/uploads/2024/10/Abstract-form.pdf, please send the completed forms to events@hg3.co.uk.
Sponsorship and Exhibition opportunities are detailed on the website.
Vaccination
I get regularly asked about vaccinations and whilst I don't work on them directly I regard them as a critical component of the healthcare system, So I've put together page on vaccinations that I hope is reasonably accessible.
https://www.cambridgemedchemconsulting.com/resources/miscellaneous/vaccination.html.
I'd be happy to add any additional information, or if anyone has a copyright free diagram of the adaptive immune system.
Proof of concept funding
UKRI have just announced funding for proof of concept to support the commercialisation of research to enable spinouts or social ventures, licensing or other commercialisation pathways. Details are here.
Applications from any disciplines are welcomed. No pre-existing UK Research and Innovation (UKRI) funding is required. The programme will not support discovery-driven research. You must be based at a UK research organisation. The full economic cost (FEC) can be up to £250,000 for 12 months duration with a minimum of £100,000 for 6 months. UKRI will fund 80% FEC.
This UKRI funding opportunity aims to de-risk the commercialisation of research. This will allow research organisations and their partners to deliver better commercialisation outcomes via the establishment of successful university spinouts or social ventures, as well as developing applicable solutions through other commercialisation routes to deliver societal and economic impacts and benefits from research.
Antiviral Competition opens Jan 13th
As part of its open science mission, the ASAP Discovery Consortium is conducting a computational methods competition encompassing several modalities critical to small molecule drug discovery. This competition will be run in collaboration with OpenADMET, which is a new ARPA-H funded project under the Open Molecular Software Foundation (OMSF).
This competition will be composed of three sub-challenges:
Ligand Poses: ASAP has produced a large volume of X-ray crystallography data over its years of operation. Along this trajectory, SARS-CoV-2 Mpro was structurally enabled much earlier than MERS-CoV. This sub-challenge will recreate that situation. Given a training set of SARS-CoV-2 Mpro X-ray structures, participants will be asked to predict poses of a test set of compounds for MERS-CoV Mpro. The crystallography experiments for this sub-challenge were performed by the University of Oxford and Diamond Light Source. See here for the crystallography conditions.
Potency: Given a training set of dose-response fluorescence potency data for both targets (SARS and MERS Mpro), participants will be challenged to predict potencies for a blind set of compounds for both targets. The assays for this sub-challenge were performed by the Weizmann Institute of Science. See here for the experimental conditions.
ADMET: This sub-challenge will consist of multiple ADMET endpoints. Participants will receive training data for all endpoints and will be asked to predict the same endpoints for a blind set of compounds. The assays for this sub-challenge were performed by Bienta.
Full details and preliminary data are available online. https://polarishub.io/blog/antiviral-competition.
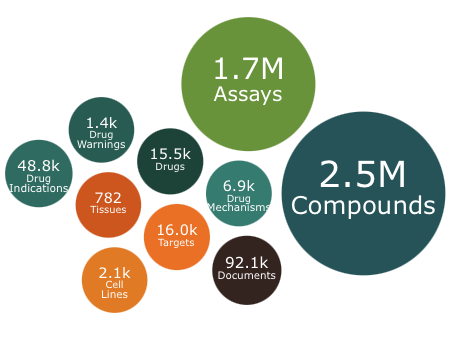
ChEMBL 35 is out
The year ends with an update to ChEMBL. This release contains 2.5 million compounds and 1.7 million assays including over 15K drugs or molecules in development.
You can download the dataset in various formats https://chembl.gitbook.io/chembl-interface-documentation/downloads.
Full details of the update are on the ChEMBL blog. https://chembl.blogspot.com/2024/12/heres-nice-christmas-gift-chembl-35-is.html.
Seasons Greetings
As many of you know I don't send Christmas cards, instead I give the monies I would have spent to MS Research. Have a great time and a successful New Year,

Comparison of protein structure prediction algorithms
The majority of drug targets are proteins and knowledge of the 3D structure of the protein can be very helpful for structure based design. Whilst the PDB contains 227,933 structures there are still a number of structures that lack structural information. In 2018 Deepmind released AlphaFold an artificial Intelligence program design to predict protein 3D structure from the amino-acid sequence DOI. Since then there have a series of updates that have included the ability to handle small molecules, co-factors, nucleic acids, protein complexes etc. AlphaFold has been used in collaboration with the EBI to create AlphaFold DB which provides open access to over 200 million protein structures, covering the human proteome and the proteomes of 47 other key organisms important in research and global health. A recent addition is Foldseek a protein structural search program that allows users to search the AlphaFold Database.

David Baker, Demis Hassabis and John Jumper were awarded the 2024 Nobel Prize for Chemistry. One half of the prize has been awarded to David Baker “for computational protein design” and the other half jointly to Demis Hassabis and John M. Jumper “for protein structure prediction.”
Whilst AphaFold gets much of the publicity, it has served to spawn a number of related programs, comparison of the different options is difficult especially when looking at the various licensing options. Fortunately, Brian Naughton has posted a very useful summary. http://blog.booleanbiotech.com/alphafold3-boltz-chai1.html.