
There are over 2 million serious adverse drug responses (ADR) per annum, ADR are 4th leading cause of death and they result in a doubling of the length of hospital stays, of the 548 molecules approved in the US between 1975 and 1999 72 were either withdrawn or had black box warnings as a result of toxicity. The toxicity can be due to drug-drug interactions but more often is thought to be due metabolic activation. In a study of compounds withdrawn due to hepatotoxicity in 5 out 6 cases there was evidence of reactive metabolites1.
Although the exact mechanism remains largely unknown, it appears to involve 2 pathways—direct hepatotoxicity and adverse immune reactions. In most instances, the liver injury is initiated by the bioactivation of drugs to chemically reactive metabolites, which have the ability to covalently react with cellular macromolecules such as proteins, lipids, and nucleic acids, leading to protein dysfunction, lipid peroxidation, DNA damage, and oxidative stress. Additionally, these reactive metabolites may induce disruption of ionic gradients and intracellular calcium stores, resulting in mitochondrial dysfunction and loss of energy production. This impairment of cellular function can culminate in cell death and possible liver failure.
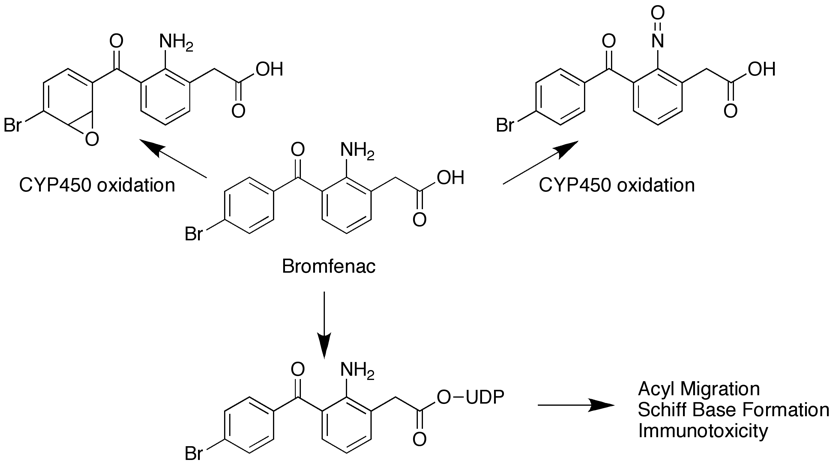
As the name implies idiosyncratic drug reaction (IDR) toxicity is not predictable based on the pharmacology of the molecule and may be a rarely seen event that might not be seen in the short-term safety studies, indeed they may be rarely seen in man. For example Bromfenac toxicity only seen in 1 in 20,000 people and yet there are at least three potential toxicophores within the molecule. Cytochrome 450 mediated oxidation of the phenyl ring leads to a reactive epoxide, or alternative oxidation of the aniline results in the nitrosomine via formation of the hydroxylamine. A third possibility is phase II conjugation leading to formation of an unstable acyl glucoronide that under goes acyl migration to reveal a reactive aldehyde. These reactive species can then react with an endogenous protein, which is then not recognised by the host and an immune response initiated.
Whilst toxicity in many organ types have been observed the majority of ADR involve the liver. Over 600 drugs have been associated with hepatotoxicity, ranging from mild, asymptomatic changes in serum transaminases, which occur at a relatively high frequency with a number of drugs, to complete hepatic failure requiring liver transplant.
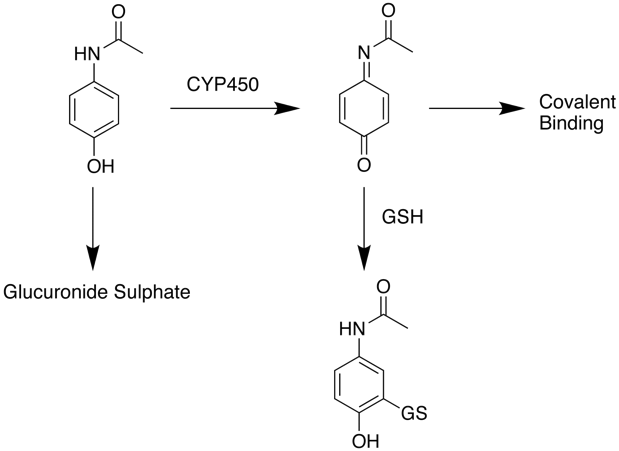
The major role of the liver is the removal of xenobiotics and it perhaps not surprising that the high concentrations of drugs found in the liver also result in high concentrations of potentially reactive metabolites. In some cases the normal route for clearance may be quite benign but at high drug concentrations an alternative toxic route may be involved. In the case of acetaminophen (paracetamol) at therapeutic doses the main route for clearance is glucuronylation and sulphation followed by renal clearance. A minor amount the parent compound is oxidised to N-acetyl-p-benzoquinoneimine and mopped up by glutathione (GSH). after overdose however this pathway is saturated and the reactive N-acetyl-p-benzoquinoneimine covalently reacts with liver proteins.
Hyman Zimmerman made the empirical observation that elevated ALT greater than 3 times the upper limits of normal (ULN) accompanied by jaundice was associated with a mortality between 5% and 50%. This observation has since been referred to as “Hy’s rule” and is currently employed by the FDA in the evaluation of hepatotoxicity for newly developed drugs. However, it must be noted that 3 × ULN of ALT levels are not necessarily predictive of overt severe liver toxicity or acute liver failure.
Strategies to avoid problems
Avoid potential toxicophores, Inherently reactive groups (quinones), Electron-rich rings (Furans), Anilines (NR2, -NHOH, NO2 etc), conjugatable group (acids, phenols). The molecule must not contain reactive groups: acrylamide, acyclic diketyl, acyl halide, aldehyde, aliphatic imine, aliphatic ketone, aryl or aliphatic nitro(so)/hydroxylamine, aliphatic (thio)ester, alkyl halide, anhydride, azide, aziridine, beta-heterosubstituted carbonyl, epoxide, halopyrimidine, hetero-allyl, iso(thio)cyanate, maleimide, Michael acceptor, perhalo ketone, phosphonate ester, phospho-, sulfonate ester, thiol, thio(urea), O-O single bonds and transition metal.There have been a number of efforts to flag potential chemical functional groups that may cause problems, these toxicophores often represent groups that might undergo metabolic activation.
Reduce partition into the liver, reduce logD.
Testing compounds for propensity to form reactive species
Whilst avoidance is a reasonable strategy it is clear many compounds have the potential for metabolic activation, however it is also apparent that not all bioactivated intermediates subsequently cause ADRs in the clinic. Variations in the electrophilicity, reactivity towards bio-molecules, detoxification pathway, and subsequent immunological recognition of the subsequent adducts must influence the outcome. It is thus important to have a strategy in place to try and assess the risk of potential adverse events and to put them into perspective with respect to the clinical exposure.
If radiolabelled compound is available then direct measurement of irreversible binding (covalent) to compound to biomolecules both in vitro and in vivo can be undertaken. It has been found that covalent binding to human liver hepatocytes was the best indicator of potential liability, but you need to take into account the likely clinical dose (see below).
In the absence of radiolabelled compound ,mass spectrometry can be used to identify glutathione conjugates or check for glutathione depletion. It should be noted that glutathione does not react with all electrophiles, hard electrophiles are better trapped with lysine or histidine. It should also be noted that not all compounds that generate glutathione adducts cause ADRs.
Several models of cell toxicity have been reported3 including a panel of immortalized human hepatocyte derived cell lines (THLE) which express (or do not express at all) high activities of individual human CYP isoforms. These toxicity in these cells discriminate between marketed drugs which cause idiosyncratic toxicity in man or not with high specificity (>99%) and good sensitivity (69%).
Comparison of HepG2 cytotoxicity in galactose vs. glucose medium has been used to assess mitochondrial impairment, a potential step in organ toxicity.
The Bile Salt Export Pump(BSEP) is responsible for pumping bile acids from hepatocytes into bile a number of drugs which cause cholestatic drug induced liver injury in man inhibit BSEP activity in vitro, therefore potent BSEP inhibition is a potential risk factor for idiosyncratic toxicity.
Cell defense systems are present that protect against chemically reactive compounds, following exposure a variety of transcription factor changes (defensive genes) have been identified, these include transcription factor AP-1, NF‑κB, nuclear factor (erythroid-derived 2)-like 1 (NFE2L1; also known as NRF1), NFE2L2 (also known as NRF2), NFE2L3 (also known as NRF3), signal transducer and activator of transcription 3 (STAT3), heat shock factor protein 1 (HSF1), pregnane X receptor, TWIST1 and E2, redox factor 1, tumour suppressor protein p53, activating transcription factor 4, CCAAT/enhancer‑binding protein 1α and the mitogen-activated protein kinases.
Potency helps
On an empirical basis, drugs administered at a total dose of 10 mg per day are unlikely to be associated with a high incidence of IDRs. This observation has been supported by recent studies2 looking at the covalent binding of 42 radiolabeled drugs in three test systems (human liver microsomes and hepatocytes in vitro and rat liver in vivo). The drugs were classified into three categories of safe (blue circles), warning (green circles), black box warning or withdrawn (red circles) based on the data given in the official documentation. It was found that covalent binding in hepatocytes was the best predictor among the three systems.
The data is shown on the plot below (created using Flot and pulling the structures from ChemSpider, with thanks to Matt for help with coding), log-normalised covalent binding is plotted against the log-normalised daily dose. All low dose compounds (log Dose less than or equal to 1) are in the “safe” category, as the daily dose increases the acceptable degree of covalent binding decreases. If you mouse over the points on the graph you should see the structure of the drug in a popup window.
Log Covalent Binding
1. Critical Reviews in Toxicology, Volume 35, Issue 4 April 2005 , pages 325 - 361
2. A Zone Classification System for Risk Assessment of Idiosyncratic Drug Toxicity Using Daily Dose and Covalent Binding, DRUG METABOLISM AND DISPOSITION Vol. 37, No. 9, 1970-1977 (2009), doi:10.1124/dmd.109.027797.
3. D.M. Dambach, B.A. Andrews, F. Moulin, New technologies and screening strategies for hepatotoxicity: use of in vitro models, Toxicol. Pathol. 33(2005) 17–26, doi:10.1080/01926230590522284.
Worth reading The Identification of Toxicophores for the Prediction of Mutagenicity, Hepatotoxicity and Cardiotoxicity
EMEA guildelines on hepatotoxicity
Lasser, K. E. et al. Timing of new black box warnings and withdrawals for prescription medications. JAMA 287, 2215–2220 (2002).
Park, et al, Managing the challenge of chemically reactive metabolites in drug development , NATURE REVIEWS DRUG DISCOVERY VOLUME 10 APRIL 2011 293.
R. A. Thompson et al, Risk assessment and mitigation strategies for reactive metabolites in drug discovery and development, Chemico-Biological Interactions (2010)
See Also CYP450 Interactions
Last Update 22 April 2011
Back to Pre-Clinical Toxicity
Back to Drug Discovery Resources