
“The single most important factor determining the likelihood of success of a project is the quality of the starting lead”, Anon
High-Throughput Screening (HTS) is one of the main sources of leads for drug discovery; the process involves evaluating a large diverse sample collection in an effort to identify compounds that possess a desired activity. In practice this resembles looking for a needle in a haystack since the vast majority of compounds in the sample collection will be devoid of the desired activity. With the increasing use of automation and library synthesis screening collections have grown in size to over a million compounds. The resulting list of “Hits” from the screen can be many 10’s of thousands of compounds and the days of a chemist simply looking through a list of structures have long gone. Nowadays the use of cheminfomatics tools has become essential for identifying the critical potential lead compounds.
“The data is not garbage, but its close” Anon
Data from high-throughput screening is generally not high quality
- Single point assay
- Sample quality variable
- Quality and diversity of Sample Collection is critical
- Compounds may interfere with the detection system
- False positives due to aggregation
- High density plates can result in cross contamination, edge effects
The objective for HTS Analysis is not to identify every active compound in the screening set, but rather to identify sufficient active series to support the active chemistry effort available.
Potential HTS analysis strategy
Cull undesirable structures
Depending on how rigorous the compilation of the screening collection has been it may be necessary to remove compounds with undesirable properties (logP, MWt, HBD, HBA,reactive groups, chelators, ligand efficiency etc.) these are described in detail in the Sample Collection section. In addition compounds that appear regularly (frequent-hitter, false positives) might also be excluded, these often derive their activity due to aggregation effects or interference with the assay technology (e.g. Calcium chelators). It may be there is a particular off-target activity that would present an insuperable problem, an early counter-screen might be available.
Whilst it might be very easy to pick out individual very potent compounds it is actually more important to identify cluster of activity. These clusters give confidence that the activity is real, may provide initial structure-activty information, and can be used to rescue false negatives (compounds that were erroneously missed in the screen). The logic behind all clustering techniques is the assumption that “similar” structures are more likely to have similar biological activity. How similarity is described can however be very different.
Substructure searching.
Perhaps the simplest technique is a medicinal chemist selecting interesting compounds and then conducting a series of substructure searches to identify compounds containing the same substructure. The substructure may be a scaffold such as a ring system, or it could be one or more functional groups that the chemist feel might represent a key recognition element or pharmacophore.
Similarity Searching
In a similar manner a chemist may select an interesting compound and search for “similar” compounds in the hit list. Usually the descriptors used to encode the molecular properties are simple 2D descriptors often based on the presence or absence of structural features to create a molecular fingerprint, searching these fingerprints is very fast and efficient and allows the user to search and browse large data sets in real time.
The disadvantage of these approaches is that they are very time-consuming and are very biased by the experience and background of the medicinal chemist. In an effort to surmount these issues automated clustering techniques have been developed in which the entire hit-list can be clustered to place similar molecules into clusters. In general these techniques focus on the molecular descriptors and not their associated biological activity.
An alternative is partitioning where all the compounds are at the top of a hierarchy and an attempt is made to divide the set into active/inactive based on the presence or absence of a particular molecular descriptor.
Clustering
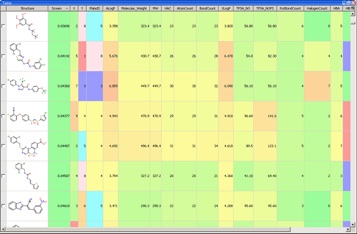
In theory any molecular descriptor can be used in clustering, in practice the use of a large number of descriptors, in particular some of the more esoteric ones, can make it difficult for a medicinal chemist to understand why compounds might be in the same cluster. It is perhaps better to stick to molecular fingerprints based on structural fragments, this usually results in clusters that chemists can see are similar. A number of clustering techniques are available but the most common are nearest neighbours, this method assigns a single compound to each of the desired number of clusters, ideally the most diverse selection. Next each compound remaining is assigned to the nearest cluster, the centroid of each cluster is recalculated as each new compound is assigned to the cluster. A useful visualisation technique is to colour code the clusters based on the average activity of the compounds in the cluster and to use a tool like Vortex to display the results. The chemist can then very easily investigate active clusters and explore possible SAR.
A particularly useful method of clustering is to cluster by maximum common substructure (MCS), most chemists when looking a set of hits will endeavor to identify a common core structure and the automated clustering by MCS provides this sort of analysis. The MCS is the largest common part between two or more compounds excluding hydrogen atoms. This technique reduces the molecules to graphs with atoms represented as nodes and bonds vertices. Owing to the NP-complete nature of the problem the difficulty increases for both larger molecules and larger data sets and so for larger data-sets it may be prohibitively time-consuming to undertake a rigorous clustering.
“The pairwise comparison of each and every compound in a set of compounds is practically infeasible, since it would involve n · ( n - 1 ) / 2 MCS calculations, where n denotes the number of compounds in the library. In the case of 1000 structures 499,500 pairwise calculations are needed. Even one such comparison is fairly complex and requires significant computation time. If, let's say the computation time of one MCS search is 10 ms on a certain hardware, the full evaluation of a 1000 library would take 4995 seconds, that is, nearly 1.5 hours (and 35 hours for 5000 molecules)” ChemAxon presentation
Fortunately a number of tools have been developed that offer faster solutions. In the approach used by ChemAxon they use similarity guided MCS, the idea being that compounds that are similar in structure are more likely to share MCS. LibMCS provides both a command line tool for setting up the calculation and a GUI for exploring the results.
Partioning (Decision Trees)
One of the potential issues with many QSAR models is an assumption that the activity of all compounds can be explained by a single (often linear) model, Recursive Partitioning methods overcome these difficulties. RP methods are able to model nonlinear relationships of almost arbitrary form, even in the presence of strong interaction between the predictors. The output of a recursive partitioning analysis is a dendrogram (tree) in which predictors are used to progressively split the data set into smaller and more homogeneous subsets. If some node in the dendrogram contains mainly active compounds, then the detailed path by which its molecules are split out provides a clue to the molecular structures that are associated with activity and perhaps a pharmacophore. Other clusters of actives in the dendrogram will have a different path and thus potentially a different pharmacophore. The ability to identify multiple independent series of hits is particularly important.
However it should be said that on very many occasions it can be very difficult to understand the model and it is treated as a black box.
The dendrogram can also be used to predict activity, a particular attractive feature is the ability to rescue false negatives. It can also be used to screen commercially available compounds.
Worth reading:- Analysis of a Large Structure/Biology Activity Data Set Using Recursive Partitioning, JCICS, A. Rusinko et al., V. 39 (6)
Selecting from Singletons
The strategies described above identify cluster of actives however in practice there is often a long tail of singletons that appear to have no similars in the screening collection and the challenge is to distinguish between novel actives and the random false positives. One approach is to use some of the clusters of active identified to generate pharmacophore queries, and to use these pharmacophore queries to mine the singletons. Any information about the molecular target can also be used such as the hydrogen bond donor/acceptor motif found in many kinase inhibitors, or the presence of an active site acid/base. If crystallographic information is available for the molecular target (or a related structure) it may be possible to use molecular docking to priortise the hits. However perhaps the most interesting are those compounds that could represent a novel pharmacophore and here I’m afraid it is down to a medicinal chemist browsing through a list of compounds together with the information described below.
As any other data?
Many compounds in the screening collection may have additional experimental data previously generated, e.g ADME data. This information could be valuable in prioritizing the hits. It is also useful to calculate some measure of “binding efficiency” this can be Ligand Efficiency (LE) potency/size, usually pIC50/number of heavy atoms, Lipophilic Efficiency (LLE) pIC50-cLogP, or perhaps a combination (pIC50-cLogP)/HAC. An alternative is to calculate the Andrew’s binding energy (P. R. Andrews, D. J. Craik and J. L. Martin, Functional group contributions to drug-receptor interactions, J. Med. Chem., 1984, 27, 1648–1657.). It might also be useful to have a chemical tractability score.
With so much data available it becomes critical to have a data visualisation tool such as Vortex available for the analysis.
Evaluating the hits
The selected hits should then be retested and titrated and a full dose response generated, and ideally a secondary screen used to confirm activity.
They should also be subjected to a quality control check to asses both purity and structural characterization. Samples would be expected to be a single major peak (>95%) by HPLC using two different columns, they would should also have the correct mass. Most of which can be automated. Compounds should also be analysed by 1H NMR.
The confirmed hit series would then be used to generate leads for the project.
Last Updated 15 Jan 2010
See also Sample Collection
See also Fragment Collection
See Also Hit to Lead
Back to Drug Discovery Resources