In vitro systems for evaluating metabolism
| Enzyme Source | Availability | Advantages | Disadvantages |
|---|---|---|---|
| Microsomes | Good, from commercial sources, many species | Inexpensive, easy storage | Contain only phase I enzymes and UDP-glucoronsyl transferase. Detergent/alamethicin addition is necessary; require strictly specific substrates and inhibitors or antibodies for individual enzymes |
| cDNA-expressed individual CYP isoforms | Good, from commercial sources, many species | Can be used in HTS, roles of individual isoforms identified | The effects of only one (or a few enzymes) at a time can be investigated |
| Immortalised cell lines | Some available | Unlimited supply, can be frozen | Modest expression levels, poorly characterised |
| Primary hepatocytes | Fresh tissue needed, available commercially | Contains full complement of enzymes and systems | Require specific techniques, enzyme levels may decrease during culture |
| Liver Slices | Fresh tissue needed, supplies variable | Contains full complement of enzymes and systems | Require specific techniques, limited viability (<24h), slow transport through tissue for some compounds. Individual variation |
Using in vitro Clearance Data
The data from in vitro systems is useful in comparing compounds but needs to be used with care when used to predict in vivo clearance. There are a number of metabolising systems that are not in the liver (see below), whilst pooling tissues reduces the errors due to inter-individual variability this may still be a concern. Systems containing only phase I systems are likely to give the poorest predictions. If partition into the liver is reduced in vivo then the in vitro results may over estimate potential clearance, compounds retained in plasma or partitioning into red blood cells would be examples. Some compounds can cause induction of metabolism.
It is perhaps better to use the in vitro data to classify compounds into low, medium or high clearance. For most indication high clearance (Rat clearance >70 uL/min/mg protein) compounds would be unsatisfactory and would be expected to have short duration. Whilst the compound may turn out to have improved stability in man the rapid clearance in a likely safety species may mean it is difficult to achieve sufficient coverage in the toxicology studies. Low clearance compounds (Rat clearance <10 uL/min/mg protein) might be expected to have long duration which might lead to accumulation after repeat dosing. There may be only a single route for metabolism which may be a concern if it is a polymorphic enzyme such as CYP 2D6 (see below).
General strategies for reducing metabolism
Reduce lipophilicity, Reduce partition into liver, CYP450 active sites, introduce heteroatoms
Deactivate aromatic rings, Introduce electron-withdrawing groups, CF3, SO2, heterocycles, F, Replace with pyridyl or other heterocycles
Block N-dealkylation, Use t-butyl (no hydrogens), or steric hinderance
Similarly O-dealkylation
Replace labile groups (Bioisosteres), Change ester to amide or heterocycle, look at ester bioisosteres
Allylic or benzylic alkyl to halide or CF3
Introduction of deuterium to exploit deuterium isotope effect
Constrain molecule, Does not fit active site or cannot adopt transition state
Block possible sites for conjugation, Avoid phenols, unhindered alcohols, carboxylic acids look at phenol bioisoteres
Check activity of metabolite/Prodrug
Examples where N-oxide is as active as parent amine
Lactone prodrug of active acid
Hydroxyisopropyl replacement for methyl
Deuterium Isotope Effect
The apparently minor change in replacing hydrogen by deuterium can have a significant effect on metabolism. Deucravacitinib is a highly selective allosteric inhibitor of non-receptor tyrosine-protein kinase 2 (TYK2) used as an oral treatment for moderate-to-severe plaque psoriasis. The deuteromethylamide suppresses demethylation via the kinetic isotope effect.

Enzymes involved in drug metabolism
| Enzyme Classes | Types of drugs metabolized | Source and distribution |
|---|---|---|
| Cytochrome P450 enzymes See Below | Practically 90% of all drug substances | Hepatocyte most versatile |
| Flavin-monooxygenases (FMO; 5 forms) | Compounds with secondary and tertiary amines or sulphydryl groups (chlorpromazine, desipramine, methimazole) | Hepatocyte most versatile and comprehensive |
| Prostaglandin H synthase (COX) | PAH-diols, aflatoxin B1, aromatic amines | Liver, kidney, bladder (microsomes) |
| Alcohol/aldehyde dehydrogenases and oxidases | Various compounds with alcohol and aldehyde functions (ethanol) also some heteroaromatic systems | Many tissues (cytosol location) |
| Monoamine oxidase (MAO) | Selegiline, moclobemide | Many tissues (mitochondrial location) |
| Esterases/hydrolases/peptidases | Compounds with cleavable ester/amide bond (procaine, succinylcholine, lidocaine) | Many tissues, including blood and blood cells |
| UDP-glucuronosyl transferases; (UGT) glucuronide conjugation, | Most drugs with suitable O-, S- and N-functional groups or produced via oxidative metabolism(morphine, diazepam paracetamol) | Subcellular systems need UDPGA; hepatocytes most versatile (microsomes) |
| Sulphotransferases (SULT); sulphate conjugation (paracetamol, methyldopa) | Phenols, alcohols, aromatic amines | Hepatocytes most versatile (cytosol); subcellular systems need 3-phosphoadenosine-5-phosphosulphate PAPS) |
| GSH transferases (GST); glutathione conjugation | Epoxides, arene oxides, nitro groups, hydroxylamines (ethacrynic acid) | Hepatocytes most versatile (cytosol, microsomes |
| Acyl-CoA glycinetransferase amino acid conjugation | Acyl-CoA derivatives of carboxylic acids(salicylic acid) | Hepatocytes (mitochondria) |
| N-acetyltransferases (NAT); acylation | Amines (sulphonamides, isoniazid, clonazepam, dapsone) | Hepatocytes (cytosol) |
| Methyl transferases; methylation | Catecholamines, phenols, amines (L-dopa thiouracil) | Various tissues (cytosol) |
Cytochrome P450 mediated metabolism
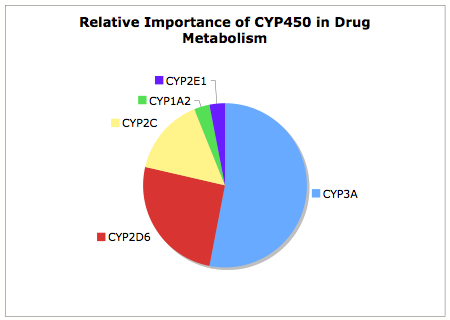
It is useful to remember of the 57 Human CYP Enzymes few have major role in drug metabolism, as can be seen from the chart below CYP3A is by far the major contributer to CYP450 mediated metabolism, this together with CYP2D6 accounts for over 75% of the CYP450 mediated metabolism. CYP2D6 shows the greatest variation in metabolic activity due to genetic polymorphism leading to individuals with normal, none or intermediate CYP2D6 activity. A patient's CYP2D6 phenotype can be determined via the administration of debrisoquine (a selective CYP2D6 substrate) and then measuring the plasma levels of the debrisoquine metabolite. The incidence of poor metabolisers varies between different populations and represents about 10% of the caucasian population. CYP2C19 expression also shows genetic polymorphism , with approximately 3–5% of Caucasian and 15–20% of Asian populations being poor metabolisers with no CYP2C19 function. Drugs metabolised solely by these isoforms would be a concern, due to the potential for overdose, and might be addressed by modification of the structure to introduce alternative metabolic pathways.

Many of the major CYP450s are now well characterised and typical substrates, inhibitors or inducers documented, these is a comprehensive list of compounds interacting with CYP450s here. Flockhart DA. Drug Interactions: Cytochrome P450 Drug Interaction Table. Indiana University School of Medicine (2007). http://medicine.iupui.edu/clinpharm/ddis/table.asp. The table below gives a summary of the major isoforms, the ones highlighted in yellow are probably the most important from a drug discovery viewpoint.
| Enzyme | Substrates (example) | Inducible | Polymorphic | Many Drug Substrates |
|---|---|---|---|---|
| 1A1 | Polycyclic aromatic hydrocarbons (benzo[a]pyrene) | Yes | No | No |
| 1A2 | Heteroaromatic amines caffeine, Theophylline, Imipramine, Propanolol, Clozapine | Yes | No | No |
| 2A6 | Small Lipophilic (coumarin) | Yes | ? | No |
| 2B6 | Small Lipophilic (cyclophosphamide) | Yes | No | No |
| 2C8 | Large Lipophilic (taxol) | Yes | Yes | No |
| 2C9 | Aromatic amides (tolbutamide), Diclofenac, Ibuprofen, Losartean | Yes | Yes | Yes |
| 2C18 | Acidic Lipophilic (phenytoin) | No | ? | No |
| 2C19 | Acidic Lipophilic (mephenytoin), Omeprazole, Citalopram, Diazepam | Yes | Yes | Yes |
| 2D6 | Basic Lipophilic (dexromethorpha), Codeine, Beta Blockers, Tricyclic Antidepressants Antipsychotics | No | Yes | Yes |
| 2E1 | Small hydrophilic (ethanol) | Yes | No | No |
| 3A4 | Large Lipophilic (midazolam), Calcium Channel blockers, Benzodiazepine, HIV Protease, HMG-CoA reductase Inhibitors, Cyclosporin, Cisapride | Yes | No | Yes |
| 3A5 | Large Lipophilic (midazolam) | Yes | Yes | No |
Predicting Metabolism
There are now a number of computational tools for predicting sites of metabolism, in general I’ve found these products very useful for identifying all potential metabolic sites, however they can over-predict and you may well find many of the potential metabolic routes have negligible contributions in vivo.
Using a CYP Inhibitor
On occasions it might be important to understand the relative importance of CYP metabolism, or to conduct an in vivo experiment with a rapidly cleared compound. In these cases it is useful to block all CYP mediated metabolism. 1-Aminobenzotriazole (ABT) is a non-isoform specific, time-dependent inhibitor of cytochrome P450 (CYP) enzymes and can be used to block most CYP mediated metabolism in vivo. It has been shown that only 5-10% of CYP activity was remaining in rat hepatocytes suspended in rat plasma following a 60 min preincubation at 2 mm ABT DOI. Atipamezole has also been shown to be a promising non-discriminative inhibitor against pan-CYP450 DOI.

In vivo utility of atipamezole was assessed by co-dosing with diclofenac in rats. At 30 mg/kg via oral, atipamezole enhanced the AUC of diclofenac by 13.1-fold and the Cmax by 5.6-fold.
Updated 25 November 2022