Solvation and Desolvation.
It is easy when considering the interactions of ligands with their protein targets to ignore the effect of solvent, hypothetical ligand binding interactions drawn on the blackboard can be misleading when trying to explain experimentally observed binding energies.
A Born-Haber cycle representing ligand binding is shown below. The 'intrinsic' free energy of binding between ligand L and protein P is represented by ΔGι whereas the experimentally observable free energy of binding is represented by ΔGexp. The terms ΔGsf and ΔGsb represent the free energy of solvation of the individual ligand and protein and the Ligand-Protein complex.

Thus the experimentally observed free energy of binding is actually
ΔGexp = ΔGi + ΔGsb - ΔGsf
Solvent contributions to changes in free binding energy arise from reorganisation of solvent-solvent and solvent-solute interactions upon binding. This mainly takes the form of desolvation of both the ligand and protein to allow their interaction. Polar ligands or side-chains on the protein can form direct hydrogen bond interactions with water. In contrast hydrophobic ligands or protein side-chains do not provide this hydrogen bonding opportunity, therefore water is believed to order at these surfaces to maximise the distance and orientation dependence of hydrogen bonds between the highly ordered network of water molecules.
Experimental Measurement of Solvation/Desolvation
It is possible to gain direct information on the role of solvent molecules in protein and protein ligand complexes using X-ray crystallography, solvent molecules that are in fixed positions relative to the protein can often be identified. The position of waters that are changing with bulk solvent are not so easily identified and in lower-resolution crystal structures, water molecules appear to be added at random positions simply to reduce the crystallographic R-factor during the refinement stage. In contrast NMR studies can provide a more dynamic picture of solvent interactions. The influence of co-solvents (Glycerol, TFE, HFIP etc.), that might be used in the experimental procedure, may have is an as yet poorly understood influence.
Quantitative information is more difficult to come by, experimental studies in the field of molecular recognition using Isothermal titration calorimetry (ITC) has provided insights in model systems.
The impact of solvation on different interaction types
The Hydrogen bond is a ubiquitous element of the recognition in biological systems, they are non-covalent, interactions between a donor hydrogen covalently bonded to some electronegative atom (typically O, N), and a acceptor electronegative atom, such as oxygen or nitrogen.
Estimation of the energetic contribution from hydrogen bonds is sometimes problematic, since in aqueous solution formation of a hydrogen bond requires the desolvation of both donor and acceptor. Whilst the strongest hydrogen bonds occur in the case of charge reinforced hydrogen bonds these are also the situation in which a higher penalty of despoliation is encountered and the net free energy gain of a stronger hydrogen bond might be reduced. In contrast hydrogen bonds for example involving C-H although weaker are much less compromised by desolvation.
Electrostatic Interactions usually involve the interaction between oppositely charged species or with dipoles, charged species in particular will incur a significant desolvation penalty.
Whilst the Halogen bond itself may be weaker than a hydrogen bond it is important to note that in contrast to the hydrogen bond where desolvation can have a significant effect on the net free energy gain, the bonding partners in a halogen bond may require minimal desolvation.
Hydrophobic interactions can be thought of as entirely driven by desolvation, a hydrophobic ligand (or surface on the binding site) disrupts the structure of bulk water and decreases entropy because of stronger bonding and ordering of water molecules around the solute, if ligand and binding site associate then some of the water molecules can be returned to bulk water. Thus the hydrophobic effect is almost entirely entropic and has been correlated with the partition between aqueous and non-polar solvents.
Aryl-Aryl Interactions are a special case, in addition to the interaction between the rings there may also be a benefit from desolvation.
There is a more detailed discussion on the molecular interactions here.
Estimation of solvent free energies
Given the critical importance of water interaction in protein folding, ligand binding, and distribution of molecules it is not surprising that considerable effort has been expended in solvent simulations. Whilst X-ray crystallography might seem an obvious source go this information in practice accurately identifying water molecules is far from trivial.
WaterMap has been used to map the locations and thermodynamic properties of water molecules that solvate ligand binding sites. GIST is another tool that uses an explicit solvent simulation to calculates thermodynamic values for the solvent located in a selected region. The 3D-RISM method produces an approximate average solvent distribution around a rigid solute. It also offers a way to compute hydration free energy (HFE). It is also orders of magnitude faster than molecular simulations. Using a protocol called WaterDock the docking tool AutoDock Vina tool can be used to predict probable binding sites for water molecules.
Examples of the impact of desolvation
Occasionally in the discussion of structure activity you will see a phrase like:-
Although this position tolerated a wide number of lipophilic substituents it was surprising that we were unable to successfully introduce simple polar substituents.
In a paper describing CDK inhibitors [DOI] the SAR in the table below was observed, whilst a range of relatively large lipophilic substituents were tolerated smaller polar substituents were not. The 4- alkoxy substituent extends into the space occupied by the ribose of ATP and does not appear to form any specific contacts with the protein. Perhaps this is a case where the energetic penalty incurred during desolvation of the polar groups compromises the overall observed binding affinity.

The targeted displacement of "unhappy" waters in the active site has been exploited on a many occasions, many waters particularly those in hydrophobic regions of the binding site do not have competitive entropy and/or hydrogen bonding as compared to bulk water. Displacement of these waters by the ligand results in significant free energy gains.
Examples of water displacement
In the design of Potent Inhibitors of Scytalone Dehydratase DOI, a key target against the pathogenic fungus, Magnaporthe grisea responsible for Rice Blast, they were able to increase inhibitory potency by synthesizing compounds were a nitrile functionality was directed into the space occupied by one of the crystallographic water molecules. Replacement of the nitrile with a hydrogen atom lowered binding affinity 100−30,000-fold.

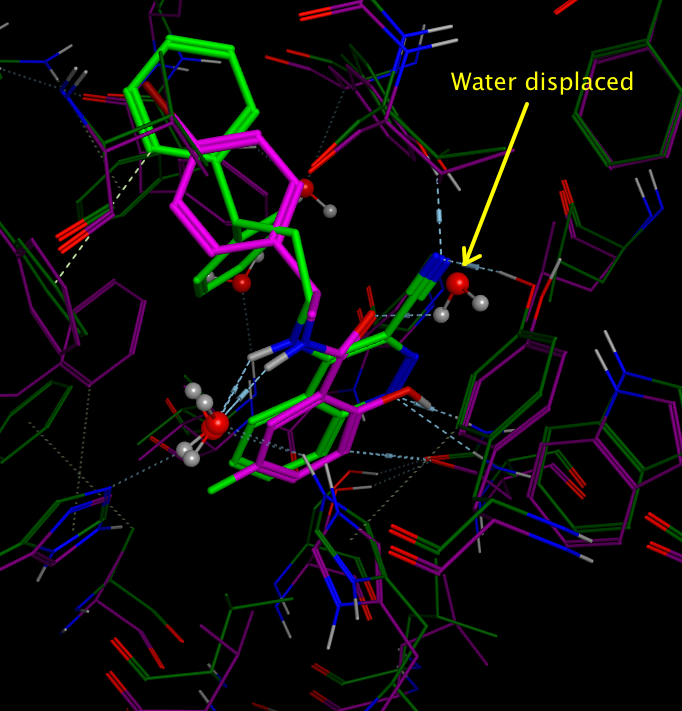
Examination of the available crystal structure 1STD identified two key water molecules involved in binding, docking studies using a quinazoline ligand suggested that the water binding to the carbonyl of the ligand shown below could be displaced. This was subsequently confirmed by crystallographic studies 3STD.

An overlay of the two crystal structures 1STD and 3STD are shown below with the water displaced by the nitrile highlighted.
In an excellent publication in Science DOI a series of nonpeptide cyclic ureas as HIV protease inhibitors were described. A fundamental feature of these inhibitors is the cyclic urea carbonyl oxygen that mimics the hydrogen-bonding features of a key structural water molecule. The success of the design in both displacing and mimicking the structural water molecule was confirmed by x-ray crystallographic studies.
HIV protease is an aspartyl protease formed as a dimer with each monomer contributing an aspartic acid to the active site. The enzyme is unique to the HIV virus and not humans, and the active enzyme is required for viral infectivity.

Examination of the crystal structure 7HVP shows a peptide inhibitor bound, with the hydroxyl bound to the two Asp25, and amides coordinated to a tetracoordinated water molecule that holds the flaps in place. Their strategy was to use rigid framework to replace structural water that is unique to all retroviral aspartic proteases, this gives selectivity but also significant entropic gain (2 kcal/mol) on liberating water to bulk solvent

As can be seen below, the carbonyl oxygen of the cyclic urea (orange) displaces the water molecule.

In a paper describing a series of 3-carbonitrile Inhibitors of Epidermal Growth Factor Receptor Kinase DOI the model suggests that with the quinazoline-based inhibitors, the N3 atom is hydrogen-bonded to a water molecule which, in turn, interacts with Thr830. It is proposed that the quinoline-3-carbonitriles bind in a similar manner where the water molecule is displaced by the cyano group which interacts with the same Thr residue.

Last update 13 May 2015